Luis Alejandro Barrera Avellaneda - La criptología de la enfermedad
Здесь есть возможность читать онлайн «Luis Alejandro Barrera Avellaneda - La criptología de la enfermedad» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на испанском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:La criptología de la enfermedad
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
La criptología de la enfermedad: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «La criptología de la enfermedad»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
La criptología de la enfermedad — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «La criptología de la enfermedad», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Algunos defectos no se pueden identificar por análisis de proteínas y hay que analizar directamente el gen, puesto que se trata de expansiones de tripletas que se pueden localizar tanto en secuencias codificadoras de los genes (por ejemplo, la enfermedad de Huntington y algunas ataxias espinocerebelosas), como en las no codificadoras (síndrome X frágil, ataxia de Friedreich) ( 32, 33).
En algunos casos los análisis enzimáticos y de ADN son contundentes para el diagnóstico, mientras que en otros no; de hecho, hay muchas mutaciones que aún se clasifican como variantes de significado incierto (VUS). Con la información clínica, la determinación de los productos que se almacenan y la respuesta a los cambios de dieta o al manejo clínico, es posible precisar el diagnóstico. En otros casos, por ejemplo cuando la información genética alterada está en el ADN no codificante, IVpor falta de un mejor conocimiento sobre esas regiones del genoma humano, aún no es posible llegar al diagnóstico ( 34).
¿Cómo se tratan los errores innatos del metabolismo?
Usando la aproximación lógica que nos enseñó Garrod, el tratamiento de los EIM se podría hacer limitando los precursores de las sustancias que se acumulan anormalmente, suministrando la proteína o enzima deficiente, reparando el gen y administrando los cofactores de las enzimas en los casos en que el EIM se deba a defectos en su síntesis o reutilización ( 35).
El primer intento de tratamiento fue la restricción en la dieta del precursor del camino metabólico que se encuentra afectado por el defecto genético. El tratamiento nutricional (restringir la ingesta de A y B en la figura 1-2) ha sido efectivo en los defectos del metabolismo de los aminoácidos, carbohidratos simples, defectos en el metabolismo del glicógeno, lípidos, vitaminas y cofactores. El tratamiento controla los síntomas, no cura la enfermedad y debe continuarse de por vida, permitiendo, en muchos casos, que los pacientes lleguen hasta la tercera edad con una vida libre de complicaciones clínicas mayores ( 36).
Una segunda aproximación de tratamiento consistiría en remplazar la proteína o enzima defectuosa (X en la figura 1-2). Para hacerlo, se requiere que esa proteína pueda ser aislada en cantidades suficientes, de manera tal que se pueda administrar y llegue al sitio de la célula donde va a actuar. Una de las dificultades es que la membrana plasmática que recubre las células impide la entrada o salida de la célula de las moléculas, especialmente las más grandes, como las proteínas, pero las células tienen transportadores que facilitan ese proceso y también las pueden internalizar por fagocitosis.
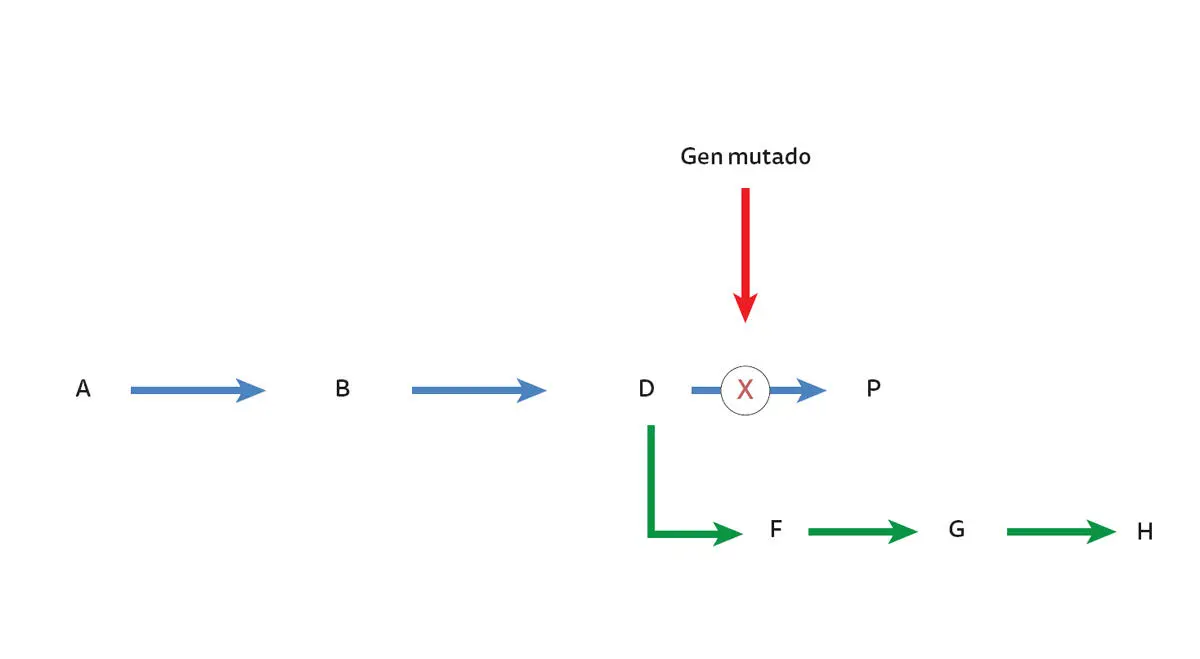
Figura 1-2. Fundamento de la terapia de reemplazo enzimático (TRE) y terapia génica (TG). En la TRE se trata de sustituir la proteína X que funciona deficientemente por daño en el gen que la codifica. La proteína usada en un comienzo era aislada de órganos o tejidos animales. Actualmente, la mayoría se sintetizan en células que se reproducen rápidamente, en las cuales se ha insertado el gen humano mediante procedimientos biotecnológicos. En la TG se busca remplazar el gen mutado por un gen normal.
Otro inconveniente es que la célula rechaza lo que no le es propio. Inicialmente se trató a los pacientes con proteínas aisladas de tejidos animales, pero bien pronto se aprendió que el organismo se defendía de esas sustancias extrañas produciendo anticuerpos muy específicos que neutralizan y facilitan su destrucción. Ese mecanismo desarrollado a través de la evolución protege a los humanos y a otros animales de sustancias foráneas. Entre más distantes son los animales en la escala zoológica, mayor es la posibilidad de que sus proteínas sean diferentes. Si se inyecta a un humano insulina de cachalote se produce mayor inmunogenicidad que con la insulina de cerdo o de ratón.
Una vez descubierta la composición y forma de sintetizar proteínas químicamente, se pensó en producir proteínas idénticas a las humanas de esa forma. Algunas proteínas pequeñas como el glucagón se han sintetizado químicamente, pero las enzimas son moléculas muy complejas, por lo que es difícil fabricarlas iguales química y biológicamente a las que sintetizan normalmente las células humanas, usando métodos químicos.
Para tratar a los pacientes era necesario buscar fuentes de proteínas tan parecidas a las humanas como fuera posible, por ejemplo, de la orina o ciertos tejidos como hígado o placenta de cerdo, de bovinos o de humanos, con la condición de que se pudieran aislar en cantidades suficientes para poder inyectar permanentemente en los pacientes, que fueran completamente puras y que no perdieran la actividad en los procesos de purificación.
Cada vez que se trata un paciente con sustancias aisladas de tejidos humanos o animales existe la posibilidad de transmitir virus u otras sustancias extrañas que originen problemas adicionales. En los años 70 se descubrió la forma de identificar y purificar los genes humanos y también la forma de introducirlos en células eucarióticas, para que ellas sinteticen la proteína de acuerdo con la instrucción programada en el gen humano, lo que en teoría debe producir una proteína idéntica a la humana, así se usen células de bacterias, levaduras, células humanas o inclusive de plantas. Pero las bacterias y las levaduras, así se les haya insertado el gen humano, producen proteínas distintas a la humana, no en la secuencia de aminoácidos, sino en las cadenas glicosiladas que hacen parte de las glicoproteínas, que son la mayoría de las proteínas de interés terapéutico ( 37- 40). Gracias a los avances en biotecnología, existen cepas de E. coli y levaduras “humanizadas” que mediante la bioingeniería se ha logrado que produzcan proteínas que tienen las mismas características de las proteínas humanas, no solo en la composición de aminoácidos, sino en otras modificaciones y adiciones que sufren las proteínas una vez que se sintetiza la cadena peptídica. Esas modificaciones hacen que las proteínas sintetizadas en los microorganismos sean virtualmente idénticas a las humanas, lo que permite “engañar” los mecanismos de defensa para que el organismo humano tolere esas moléculas sintetizadas en otras especies y puedan ser usadas en el tratamiento de los errores innatos del metabolismo ( 41, 42).
Si la proteína se inyecta por vía intravenosa, a su paso por los diferentes tejidos es captada por todas las células que tienen receptores para lo residuos glicosilados o alguna otra forma de reconocerlas, e internalizarla. Por lo tanto, las proteínas para uso terapéutico deben tener señales de direccionamiento que les permitan llegar al órgano que las necesita y no se extravíen en aquellos en los cuales no se requieren ( 43).
Los organelos de las células (el núcleo, la mitocondria, los lisosomas, los peroxisomas) tienen membranas que los recubren, por lo que la proteína exógena debe atravesar una o varias membranas para llegar al sitio donde actúa y arribar intacta en cuanto a su función. Entonces, el desafío es direccionar esa proteína sin que sea neutralizada por nuestros mecanismos de defensa antes de llegar al sitio que queremos reparar. Sin entrar en detalles, adelantamos que usando proteínas exógenas purificadas, solo ha sido posible tratar, con cierto éxito, cerca de una docena de las enfermedades lisosomales (ver capítulo 11). Los defectos lisosomales por razones estructurales y fisiológicas de la célula son los más fáciles de tratar; aún no hay terapia de reemplazo enzimático para los otros EIM, en algunos casos por complejidades metodológicas, en otros por ser enfermedades muy poco frecuentes que no generan suficiente interés para desarrollar terapias que sean rentables económicamente para una industria acostumbrada a grandes negocios y ganancias, lo que pone de presente las enormes limitaciones que existen y la difícil tarea que hay por delante en cuanto a desarrollo de terapias para enfermedades raras.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «La criptología de la enfermedad»
Представляем Вашему вниманию похожие книги на «La criptología de la enfermedad» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «La criptología de la enfermedad» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.