Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
Здесь есть возможность читать онлайн «Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
The first book dedicated to the emerging field of physiologically based pharmacokinetic modeling (PBPK) Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulations: Principles, Methods, and Applications in the Pharma Industry
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry, Second Edition
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
The clearance ( CL ) of many low hepatic‐extraction drugs is limited by protein binding. Only the unbound drug is available for glomerular filtration and therefore for renal elimination. An increase in unbound drug concentration due to a reduced plasma protein binding will enable higher tissue distribution and higher CL . However, since the half‐life of a drug is directly proportional to the distribution volume and inversely proportional to CL , there is no net effect on the half‐life. Thus, changes in plasma protein binding of a drug are not likely to be clinically relevant (Benet and Hoener, 2002) except in the following cases:
1 The drug is >98% bound to plasma proteins. In this case, even a small shift in plasma protein binding can have a substantial effect on the clearance but less so on the distribution volume, thus temporarily altering the unbound drug concentrations.
2 The drug has high hepatic extraction. The clearance of such drugs will be dependent only on the hepatic blood flow rate and not on the product of fup × CLint . Thus, an increase in distribution volume is not sufficiently compensated for by an increase in CL, leading to a temporary increase in unbound drug concentrations.
3 There is a rapid equilibrium between drug concentration and pharmacological response (e.g., lidocaine with a PK‐PD equilibration time of two minutes) compared to the time required for the body to regain equilibrium (about 30 minutes). Many anti‐arrhythmic drugs and anesthetics require only a short time for a change in concentration to cause a change in drug effect. In these cases, the response is sensitive to small transient changes in unbound drug concentrations.
Differences in unbound drug concentrations discussed in (i) or (ii) will have a greater impact on a drug with a narrow therapeutic window/safety margin. Scaling of pharmacokinetic (PK) parameters like clearance or volume of distribution or translation of pharmacodynamic properties (Mager et al., 2009) from preclinical species to man should always be done with the unbound parameters. Any comparisons/correlations of PK parameters should also be done with unbound values.
Some drugs also bind to and distribute into erythrocytes, the main drivers being lipophilicity, pKa and active uptake into the erythrocytes. Binding sites within erythrocytes are hemoglobin, proteins like carbonic anhydrase, and plasma membrane. The blood–plasma concentration ratio ( R ) of a drug is a measure of its binding and distribution to erythrocytes relative to plasma. A compound having a similar extent of binding to the constituents of erythrocytes and plasma has a blood–plasma ratio of 1. Acids tend to have R values of around 0.5 and rarely exceed 1, and bases tend to have a higher range of values, often exceeding 1 while neutrals and ampholytes have values of around 1 (Hinderling, 1997). Uchimura et al. (2010) describe several methods to determine R . Commonly, it is determined by measuring the concentrations of 14C‐labelled drug in erythrocytes ( C e) and plasma ( C p) in freshly collected blood. Then, knowing the hematocrit, H (the volume fraction of blood occupied by erythrocytes), R is obtained as follows:
(1.21) 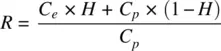
According to Equation 1.21, considering an average H of 0.45, the minimum value of R can be 0.55 which corresponds to no distribution into erythrocytes. However, there is no upper limit. For tacrolimus, R is as high as 55 and exhibits concentration dependence (Jusko et al. 1995). R can also be predicted (Paixão et al., 2009).
Partitioning can be fast for some drugs and distribution equilibrium is reached within a few seconds to minutes. However, many drugs with primary amine groups show delayed equilibrium probably due to the formation of Schiff bases with membrane fatty acids and aldehydes. While the displacement of the plasma–protein–bound drug to the unbound is rapid (except for protein molecules), displacement of erythrocyte bound drug is relatively slow. For acids with high plasma protein binding, distribution into erythrocytes can significantly affect its distribution volume, as other tissue compartments are not as significant. If blood–plasma concentration ratios exceed 1, as is the case for lipophilic bases, then plasma clearance significantly overestimates blood clearance and could even exceed hepatic blood flow. This is because the concentrations measured in plasma will always be much smaller compared to that measured in whole blood. This is due to greater distribution into the erythrocytes when R is >1. Thus, blood clearance is related to plasma clearance and blood–plasma ratio by the following equation:
(1.22a) 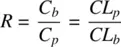
Similarly, plasma and blood volume are related by
(1.22b) 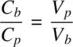
1.2.4 Hepatic, Renal, and Biliary Clearances
The CL estimated from dose and AUC ( Equation 1.10) is the overall clearance of the drug from blood, also called the total clearance. Several clearance pathways could contribute to the total clearance of a drug. Hepatic metabolism, renal, and biliary pathways are some of the clearance routes available for a xenobiotic. The most common route is hepatic metabolism. A drug may utilize one or more clearance pathways depending on its physicochemical properties. The total clearance of a drug is the sum of its hepatic ( CL H), renal ( CL R), and biliary ( CL B) clearances.
(1.23) 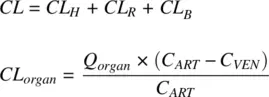
CL organis the clearance from an eliminating organ, Q organis the blood flow rate to that organ, C ARTand C VENare the arterial and venous concentrations. ( Q organ× C ART− Q organ× C VEN) is the rate of elimination from that organ.
1.2.4.1 Hepatic Clearance
The liver is the most important eliminating organ, where phase I and phase II metabolizing enzymes convert low molecular weight drugs into more hydrophilic compounds with greater molecular weight which can enter bile or undergo renal elimination. About 40 human CYP450 genes have been cloned and classified according to sequence homology. Of these, only 3 CYP450 families and <12 unique enzymes play a substantial role in the hepatic metabolism of drugs in humans. The rate of such an enzyme driven biotransformation reaction, v , depends on the free concentration of the drug, C , according to the Michaelis–Menten equation:
(1.24) 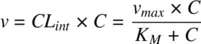
CL intis the intrinsic clearance of the drug, v maxis the maximum velocity of the reaction, and K Mis the Michaelis–Menten constant ( Figure 1.3). When the therapeutic concentration range is low relative to K M(true for many drugs), Equation 1.24becomes:
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»
Представляем Вашему вниманию похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.