Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
Здесь есть возможность читать онлайн «Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
The first book dedicated to the emerging field of physiologically based pharmacokinetic modeling (PBPK) Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulations: Principles, Methods, and Applications in the Pharma Industry
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry, Second Edition
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
A metabolite of the drug can also be emptied into bile and into duodenum Figure 1.5. Certain phase II metabolites such as glucuronides are emptied into bile and into duodenum, get reconverted to the parent drug by the gut microflora in the distal intestine and are reabsorbed and recirculated. Although the pharmacokinetic profile is very similar to that associated with parent EHR , the measured C bilefor the parent drug in this case would be low, allowing one to distinguish between parent and metabolite EHR .
1.2.5 Extravascular (Subcutaneous, Intramuscular, and Per Oral) Absorption
Other than IV administration, all other routes require the drug to be absorbed into the capillaries that surround the site of administration. The rate of absorption of a drug in solution administered by IM and SC routes is limited by the perfusion to the tissue. It also depends on the type of tissue at the site of administration – the density, vascularity, and the fat content. The IM route for example has higher rate of absorption compared to the SC because of lesser fat and greater vascularity of the dense muscles. The muscle tissue has a greater blood supply than the tissue just under the skin and can hold a larger volume of medication than subcutaneous tissue.
Oral drug absorption refers to the transport of drug molecules across the enterocytes lining the gastrointestinal (GI) tract into the venous capillaries along the gut wall. The rate of oral drug absorption depends on several physiology‐, drug‐, and formulation‐dependent factors such as gastric emptying rate, intestinal motility, porosity of tight junctions, luminal and mucosal enzymology, carrier and efflux transporters, small‐intestinal secretions (bile and digestive enzymes), regional differences in pH, membrane permeability, solubility, and dissolution rate. An orally absorbed drug is subjected to first pass metabolism in the gut and liver before it is available in systemic circulation.
PK profiles from all extravascular routes are characterized by a maximum systemic concentration ( C max) and t max, the time at which the C maxis achieved ( Figure 1.6a). Only a fraction of the administered dose is available for systemic circulation. This fraction is called bioavailability ( F ) of the drug.
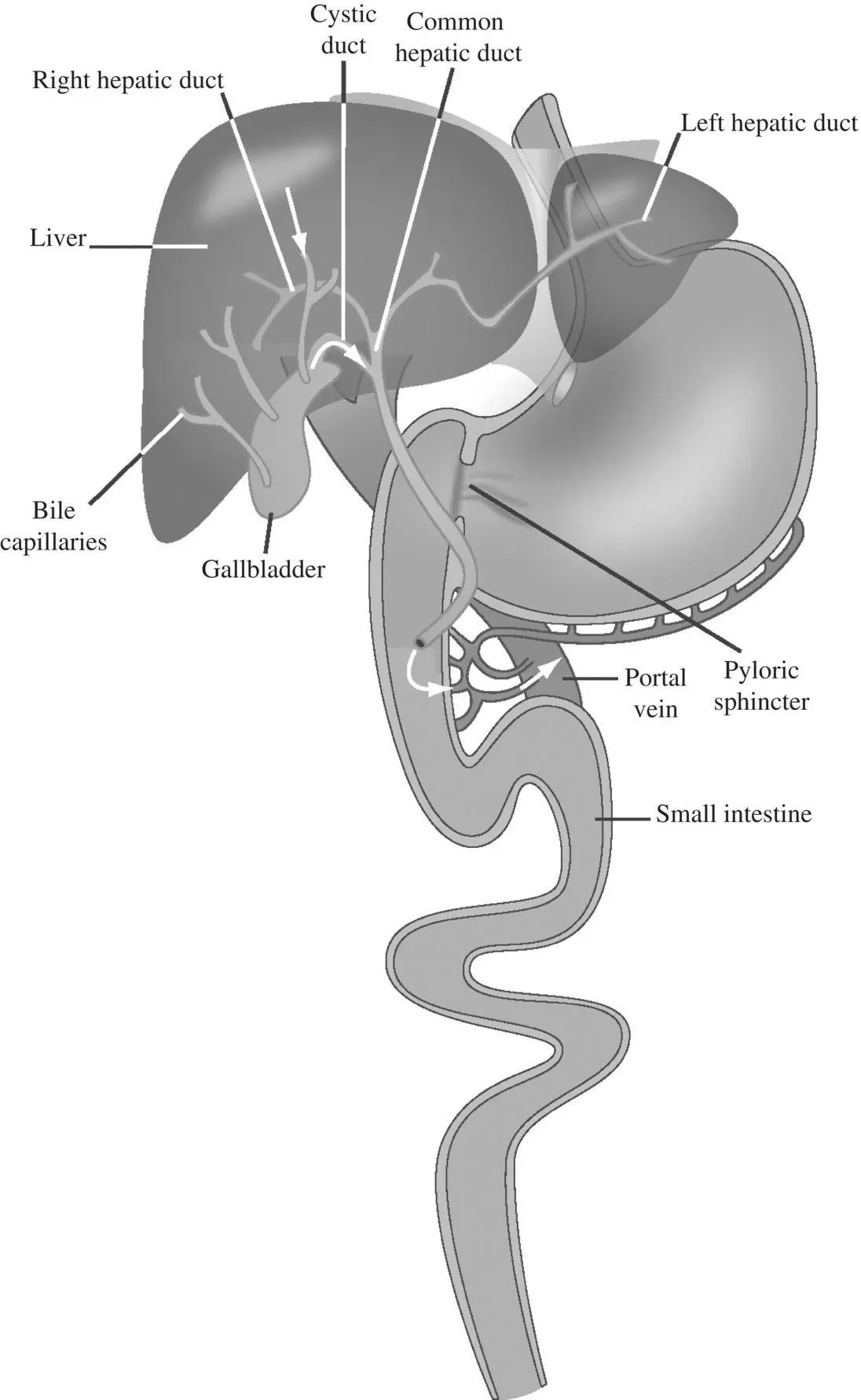
Figure 1.5. Entero‐hepatic recirculation of a parent drug or a metabolite.
Considering first‐order absorption, the rate of change of the amount of drug in the body following oral administration is given by the rate of drug absorbed into systemic circulation ( 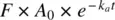 is the amount available at any time for absorption) minus the rate of drug elimination from systemic circulation as shown in Equation 1.32a:
is the amount available at any time for absorption) minus the rate of drug elimination from systemic circulation as shown in Equation 1.32a:
(1.32a) 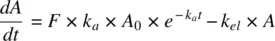
Integrating 1.32a, and dividing both sides by volume,
(1.32b) 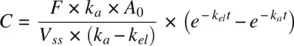
The oral bioavailability of a drug can be estimated from the areas under the PK profiles of intravenous and oral doses, by assuming that the hepatic clearance does not vary between the IV and oral routes. When the oral and IV doses are the same (see Figure 1.6b, c),
(1.33) 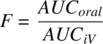
When the oral and IV doses are different,
(1.34) 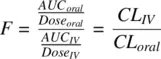
Therefore,
(1.35) 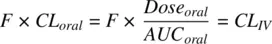
Similar Equations 1.32–1.35 apply for other extravascular routes also. If absorption from the site of administration is fast, the terminal slope of the semilogarithmic concentration‐time profile is defined by drug elimination rate. It is therefore parallel to the terminal slope of IV profile (see Figure 1.6b, c). It is then possible to extract the absorption rate constant and the elimination rate constant by the curve stripping method illustrated in Figure 1.6d. Alternatively, t maxmay be calculated ( t max= ln( k a/ k el)/( k a− k el)), assuming that contributions from processes other than absorption to t maxare negligible. In SC and topical administrations, drug absorption is slow, and the absorption rate constant, k a, is much slower than the k el(elimination constant) ( Figure 1.6e). This is also referred to as flip‐flop kinetics . The PK profile in these routes is sometimes characterized by a lag time t lag. Flip‐flop kinetics and lag time can also occur when the oral route of administration is used, if gastric emptying is delayed for some reason (fed state, elderly, drug‐induced etc.).
The oral bioavailability of a drug is given by
(1.36) 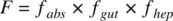
where f absis the fraction of dose absorbed, f gutis the fraction escaping gut extraction, and f hepis the fraction escaping hepatic extraction. If the IV PK profile is available, knowing the hepatic clearance, hepatic extraction, E H, can be calculated using Equation 1.26. f hepis related to E Hby
(1.37a) 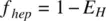
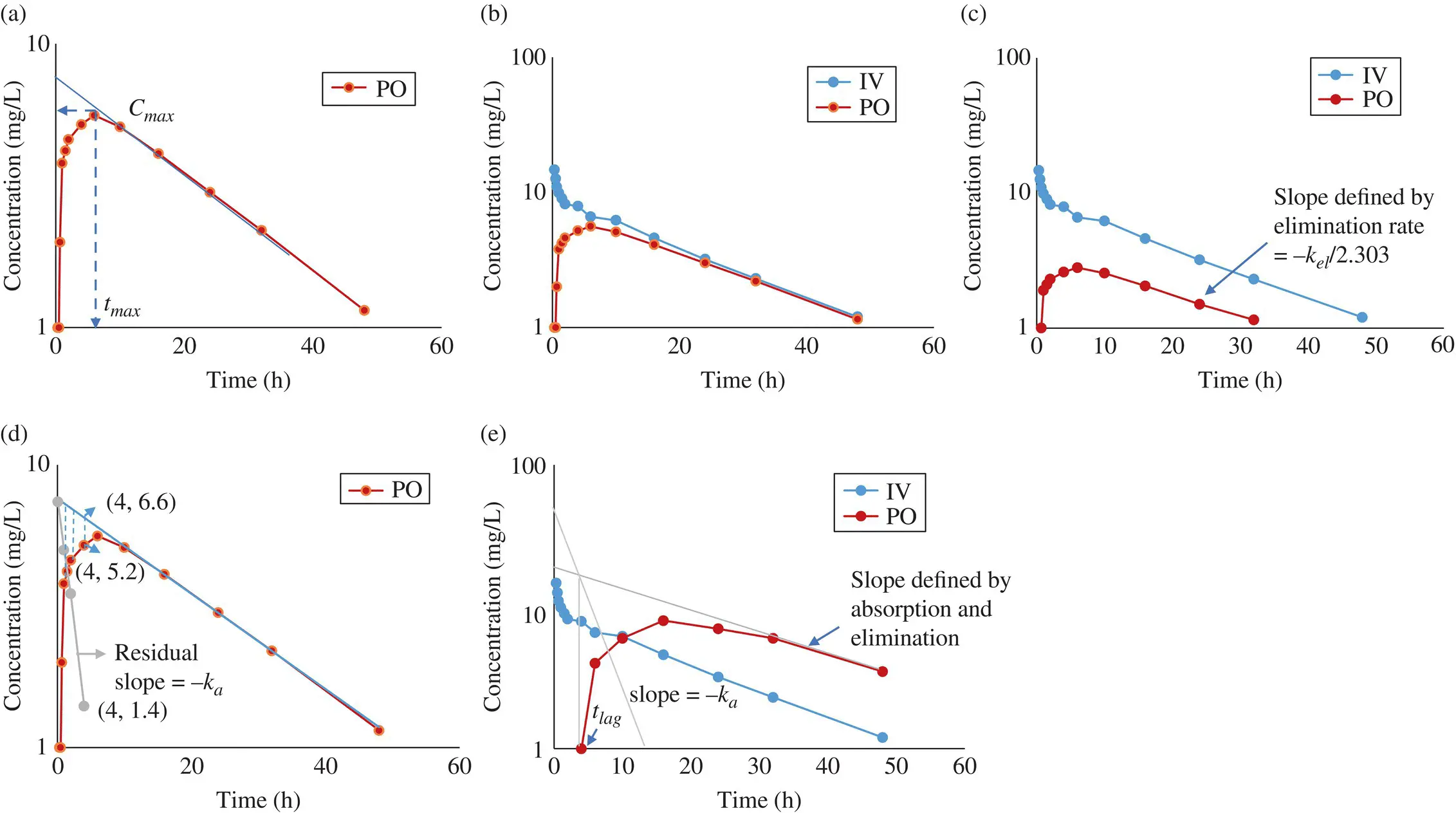
Figure 1.6. Oral pharmacokinetic profiles. (a) Maximum systemic concentration ( C max) and time at which C maxis achieved ( t max) following oral administration of a drug. (b) AUC for an orally administered drug that is nearly half of the AUC following IV administration of the same dose of that drug, indicating that the drug has about 50% oral bioavailability. (c) Area under the curve ( AUC ) for an orally administered drug that is nearly the same as the AUC following IV administration of the same dose of that drug, indicating that the drug has high oral bioavailability. (d) Curve stripping: Absorption rate constant k ais obtained from the residual slope of the straight line constructed by plotting the differences between the back‐extrapolated terminal slope and the observed curve in a semilogarithmic plot (illustrated with one of the four points plotted). (e) Flip‐flop kinetics with a lag time, t lag. Using the curve stripping method described in (d), the t lagand the k acan be obtained as shown.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»
Представляем Вашему вниманию похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.