Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
Здесь есть возможность читать онлайн «Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
The first book dedicated to the emerging field of physiologically based pharmacokinetic modeling (PBPK) Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulations: Principles, Methods, and Applications in the Pharma Industry
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry, Second Edition
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
(1.25) 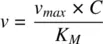
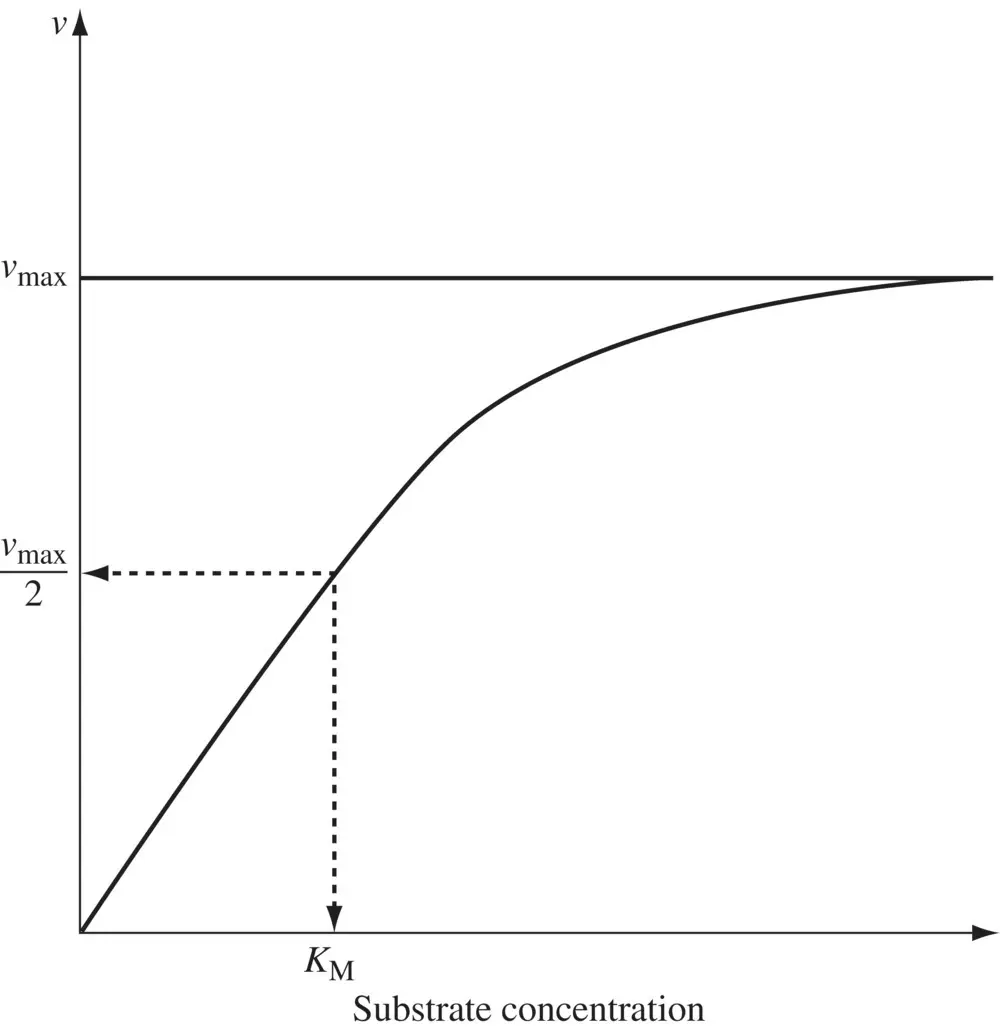
Figure 1.3. Rate of an enzyme‐catalyzed reaction as a function of substrate concentration.
Equation 1.25suggests that the rate of a metabolic reaction varies linearly with the drug concentration at concentrations not exceeding K M. CL intthen equals the ratio of v maxto K Mand is independent of the drug concentration. However, when drug concentrations are comparable with or exceeds K M, which can be the case at high drug doses, CL intis dependent on C (see Equation 1.24). The greater the drug concentration, the greater the extent of enzyme saturation and smaller the value of CL int. A maximal rate of metabolism v maxis reached at concentrations much higher relative to K M. Under these conditions, zero‐order kinetics is said to prevail, and a constant amount of drug is eliminated per unit time, independent of the amount of drug in the body. Well‐known examples of drugs exhibiting nonlinear clearance include phenytoin, ethanol, methyl salicylate, and theophylline (in some individuals). Apart from CL int, the hepatic clearance CL His dependent on how fast the drug is delivered to the enzymes in the hepatocytes determined by the total blood flow rate to the liver, Q LI. A basic tenet of pharmacokinetics is that only the fraction of drug that is not bound to blood, f ub, is available for distribution into tissues and for clearance. The dependencies of CL Hon CL int , Q LI, and f ubare encapsulated in the well‐stirred model, in which drug concentration in the liver is assumed to be uniform throughout the tissue. According to this model, the hepatic clearance from blood, CL H, is given by:
(1.26) 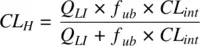
Equation 1.26shows that when the product f up× CL intis high compared to Q LI,then CL His simply determined by the blood flow rate to the liver. The drug clearance is limited by the rate at which it is delivered to the drug metabolizing enzymes. On the other hand, when the product f up× CL intis low compared to Q LI,then the hepatic clearance linearly varies with the product of fraction unbound in plasma and the intrinsic clearance.
The fraction of drug unbound in blood, f ubis related to f upand R as follows:
(1.27) 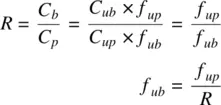
1.2.4.2 Hepatic Extraction
The hepatic extraction ratio of a drug is obtained by taking Q LIto the left‐hand side in Equation 1.26
(1.28) 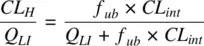
For high CL intcompounds, the displacement of the drug from plasma proteins is rapid and equilibrium cannot be established between concentrations of the bound and unbound drug in blood ( C b, C u,b) and in liver ( C u,liver). This can be represented as:
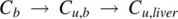
For low CL intcompounds, there is equilibrium between the bound drug and unbound drug in blood and liver and only the C u,liveris available to the drug metabolizing enzymes. The equilibrium between the different drug concentrations is shown below:
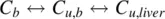
1.2.4.3 Renal Clearance
Hydrophilic drugs can get eliminated in the urine, unchanged. For example, renal elimination is the predominant route for about 60% of anti‐infection compounds (Varma et al., 2009). The fraction of the dose excreted in the urine unchanged ( f e) is
(1.29) 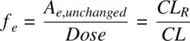
where A e,unchangedis the amount of drug excreted in the urine unchanged. Therefore, CL Ris
(1.30) 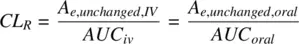

Figure 1.4. Renal elimination of a drug: glomerular filtration, active tubular secretion, and tubular reabsorption. Lipophilic drugs are readily reabsorbed, making renal elimination an important route only for hydrophilic drugs.
CL R= f ub× GFR , where GFR is the glomerular filtration rate , for a drug lacking tubular active secretion or tubular reabsorption ( Figure 1.4). GFR reflects passive diffusion of a drug through the glomeruli. Active tubular secretion is evident if CL R> f ub× GFR and tubular reabsorption is apparent when CL R< f ub× GFR . Reported values of GFR are measured with endogenous filtration markers like creatinine, which is freely filtered and secreted (15%) in the proximal tubule. However, since the synthesis and blood concentration of creatinine are influenced by several factors including age, sex, ethnicity, muscle mass, and chronic illness, other markers such as serum cystatin C, 51CrEDTA or inulin are employed. Drugs cleared exclusively in the kidney (such as hydrophilic acids and bases with high PSA and rotatable bond count), generally tend to have low rates of renal clearance. This is due to limited glomerular filtration and in some cases, absent active secretion. Apart from low clearances, these compounds are also unaffected by CYP‐related issues such as polymorphisms , drug–drug interaction (DDI) and reactive metabolites.
1.2.4.4 Biliary Clearance
Amphiphilic compounds (compounds with both acidic and basic groups), with molecular weights >350 Da also have the possibility of being actively transported into the bile and excreted via faces. Biliary clearance can be estimated by determining the concentration of a drug in the bile ( C bile) collected from a bile‐duct cannulated preclinical species.
(1.31) 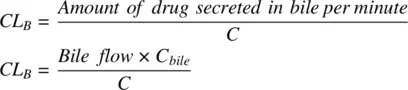
The parent drug in bile is emptied into the duodenal section of the small intestinal tract via the sphincter of Oddi and may be reabsorbed back into the portal vein as it transits down the intestine. This is called the enterohepatic recirculation ( EHR ). EHR contributes to an increased half‐life of a drug. Some phase II conjugates are also secreted into bile and converted back to parent in the distal small intestine and reabsorbed as parent.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»
Представляем Вашему вниманию похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.