Handbook of Aggregation-Induced Emission, Volume 1
Здесь есть возможность читать онлайн «Handbook of Aggregation-Induced Emission, Volume 1» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Handbook of Aggregation-Induced Emission, Volume 1
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Handbook of Aggregation-Induced Emission, Volume 1: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Handbook of Aggregation-Induced Emission, Volume 1»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
A thorough introduction to the mechanistic understanding of the importance of molecular motion to aggregation-induced emission An exploration of the aggregation-induced emission mechanism at the molecular level Practical discussions of aggregation-induced emission from the restriction of double bond rotation at the excited state, and clusterization-triggered emission Perfect for academic researchers working on aggregation-induced emission, this set of volumes is also ideal for professionals and students in the fields of photophysics, photochemistry, materials science, optoelectronic materials, synthetic organic chemistry, macromolecular chemistry, polymer science, and biological sciences.
Handbook of Aggregation-Induced Emission, Volume 1 — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Handbook of Aggregation-Induced Emission, Volume 1», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
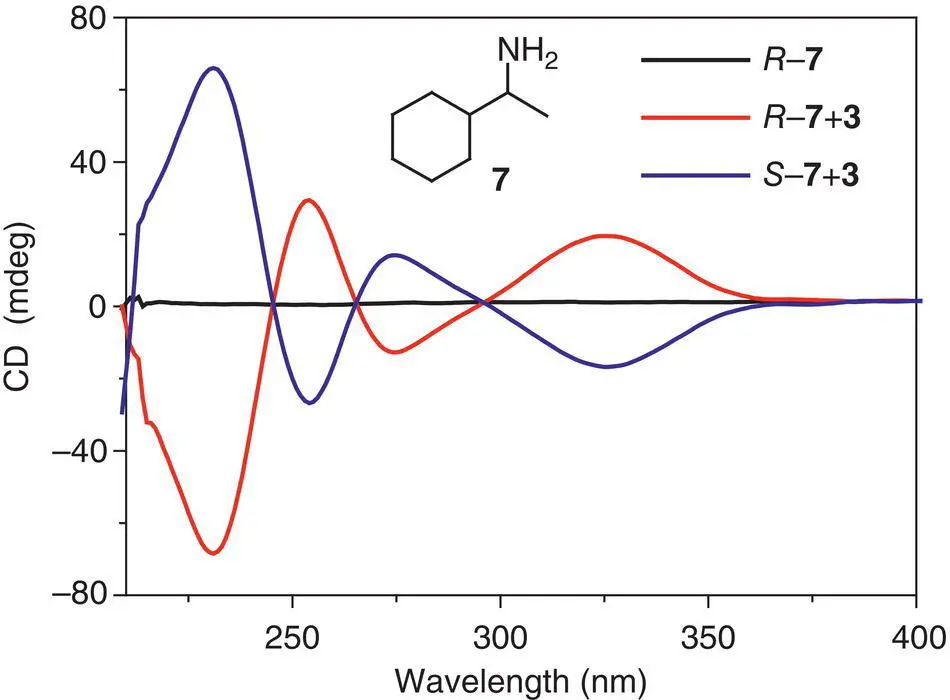
Figure 3.16 CD spectra of a mixture of TPE dicycle 3and enantiomer of α‐methylbenzylamine 7in the presence of acetic acid in 1,2‐dichloroethane.
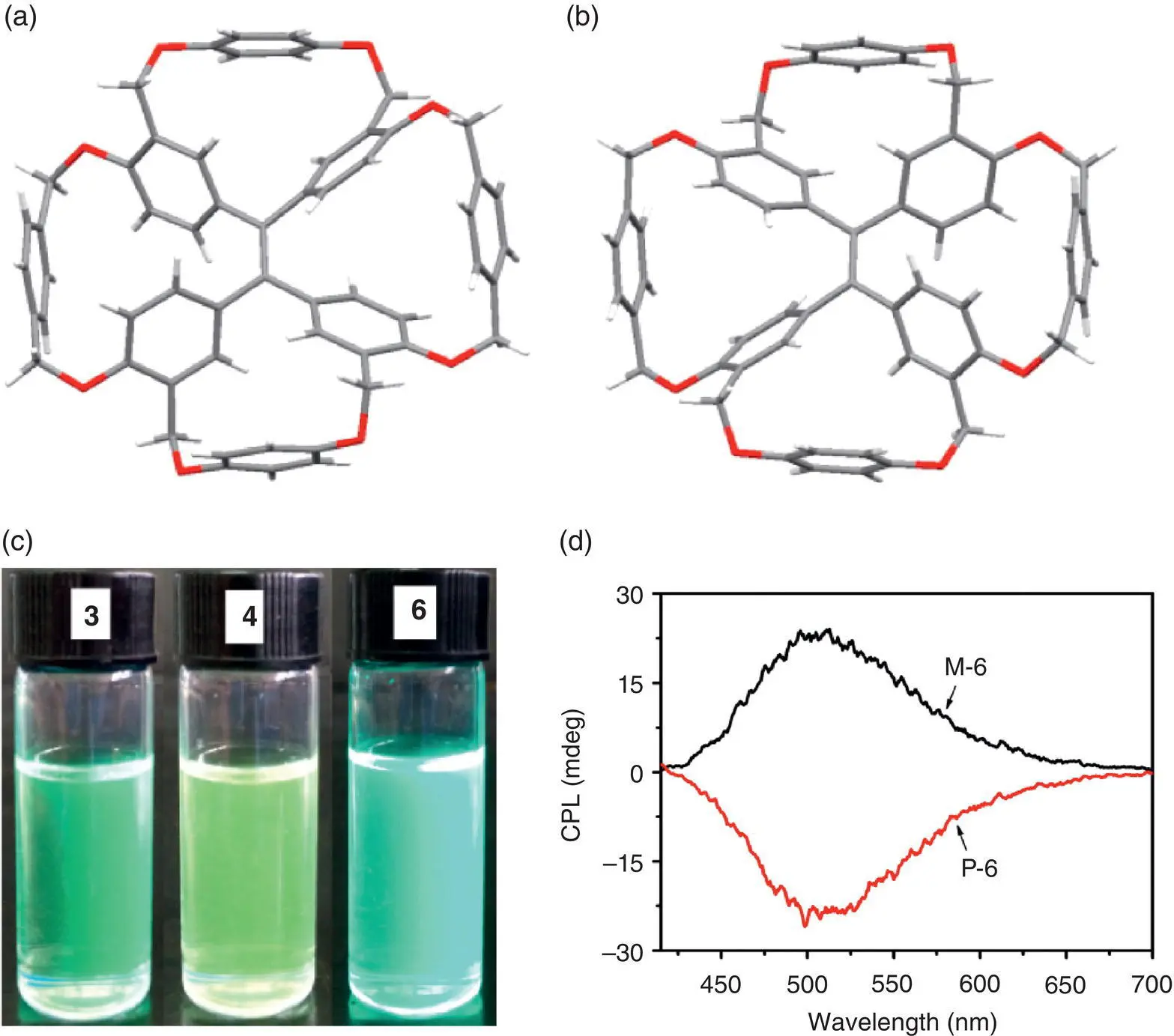
Figure 3.17 The crystal structures of M ‐ 6(a) and P ‐ 6(b); (c) photos of 3, 4, and 6in THF solution under a 365‐nm UV light and (d) CPL spectra of M ‐ 6and P ‐ 6in THF (1.0 × 10 −3M).
Source: Reproduced with permission from Ref. [43]. Copyright 2016, American Chemical Society.
In order to further disclose the important contribution of the RDBR process to the AIE effect, gem ‐TPE dicycles 7and 8and even the typical isomers cis ‐ and gem ‐TPE dicycles 9and 10were designed and synthesized in Zheng’s group (see Figure 3.18) [44]. Their configuration had been confirmed by the crystal structure. By comparing the fluorescence intensity of these isomers, the effect of groups, atoms, and the bridge chains on the fluorescence could be excluded. Therefore, more direct and more exact RDBR evidence could be furnished.
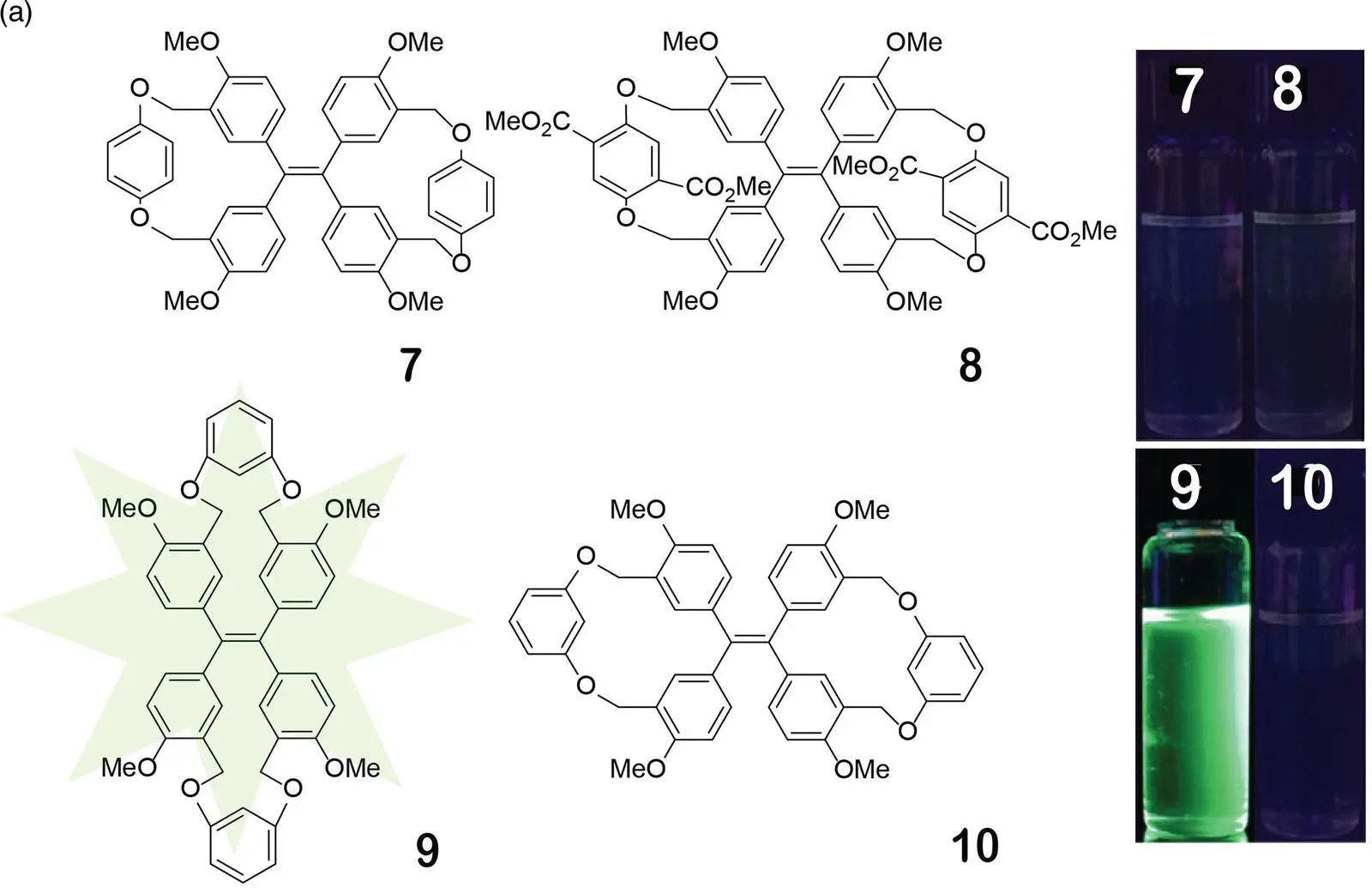
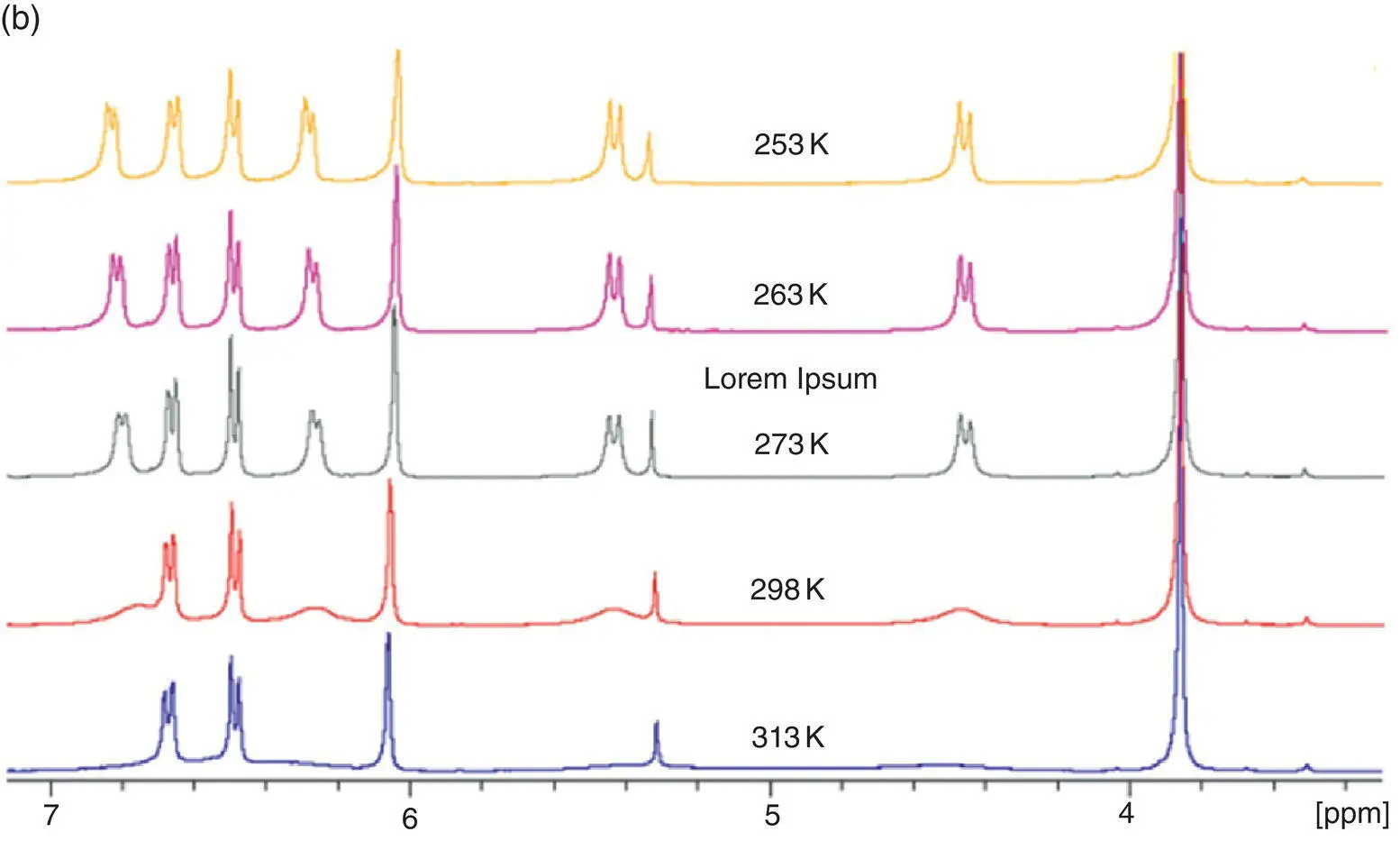
Figure 3.18 (a) Structures of TPE dicycle isomers 7– 10(left) and photos of their solution in THF (1.0 × 10 −4M) under a 365‐nm UV light (middle). (b) The change of 1H NMR spectra of 7in CDCl 3with temperature.
Source: Reproduced with permission from Ref. [44]. Copyright 2018, American Chemical Society.
As expected, while cis ‐TPE dicycles 3, 4, and 9emitted a strong fluorescence, the gem ‐TPE dicycles 7, 8, and 10had no emission in THF under an irradiation of a 365‐nm UV light. The fluorescence quantum yields of the cis ‐TPE dicycles 3, 4, and 9were 24, 49, and 22%, while those of the gem ‐TPE dicycles 7, 8, and 10were 3.1, 5.7, and 1.9%, respectively, in THF at 25 °C. Compared with the gem ‐isomer 10, the cis ‐isomer 9displayed an increase of fluorescence intensity by 11.6‐fold. From the 1H NMR of the gem ‐TPE dicycle 7at different temperatures, the phenyl ring of the TPE unit was fixed and failed to rotate due to linkage of the bridge at the m ‐position of the phenyl rings. However, the bridging unit 1,4‐benzenedioxymethyl was rapidly rotating at 25 °C because the proton peaks of both the 1,4‐benzenedioxy and methylene unit were very wide and flat. When the temperature was lowered to 0 °C, these proton resonance signals became sharp and well resolved, indicating that the free rotation of these bridge units was also restricted. However, the Φ fof 7was only increased to 9.0% at 0 °C and was still much less than that of cis ‐isomers. Compared with the cis ‐isomer, the fluorescent decrease of the gem ‐isomer in solution only came from free rotation of the double bond at the excited state after all other intramolecular rotations had been restricted (see Figure 3.18right).
Time‐resolved fluorescence decay of TPE dicycles 3, 4, and 7–10disclosed that the fluorescence lifetime was in the range of 6.9–14 ns in THF and 13–19 ns in suspension for these TPE dicycles. In suspension, their fluorescence lifetime was always larger than that in solution. This demonstrated that there was further restriction of intramolecular rotation in solid state. By making use of the fluorescence quantum yields and lifetime values, the radiative ( k f) and nonradiative ( k nr) rate constants of 3, 4, and 7–10( k f, k nr(ns −1)) could be calculated. The k fand k nrwere 0.035 and 0.112 for 3, 0.051 and 0.053 for 4, 0.004 and 0.126 for 7, 0.004 and 0.067 for 8, 0.023 and 0.080 for 9, and 0.002 and 0.103 for 10. And ratios of k nrvs. k ffor 3, 4, and 7–10were 3.20, 1.04, 31.5, 16.8, 3.48, and 51.5, respectively. The k nr/ k fratio from gem ‐isomers was always much larger than that from cis ‐isomers, demonstrating a more nonradiative process from gem ‐isomers. This nonradiative process should be mainly ascribed to the double bond rotation.
If the double bond rotated at the excited state, one intermediate state in which sp 2‐hybridized orbital planes of two carbons are vertical instead of coplanar should exist. This twisted state of the double bond should be able to be observed by femtosecond transient absorption spectra because of the decreased conjugation with one another. It was true that two excited‐state absorption (ESA) bands, which were located at <460 and >600 nm, respectively, were observed. The former should come from a twisted excited double bond and the latter came from a planar double bond at the excited state, corroborating the occurrence of the double bond rotation. Surprisingly, the two ESA bands existed both in gem ‐isomers and in cis ‐isomers. Two typical time components τ 1and τ 2from the dynamic decay process were obtained. By the global analysis, the first component τ 1representing the rise component of ESA and associated with the geometry relaxation from the Franck–Condon (FC) configuration was the negative amplitude. The second component τ 2represented the nonradiative internal conversion (IC) decay of the excited state. From time‐resolved spectra, an obviously growing process and the increase ending up in less than 20 ps were observed for the species at short wavelength, suggesting that the planar excited state could be transformed into the twisted excited state. Therefore, the double bond rotation at the excited state occurred for all the TPE dicycles.
However, there was an obvious difference of the double bond rotation between the cis ‐ and gem ‐isomers. As an index of the rotation, the component τ 1was 6 ps for gem ‐ 10and obviously shorter than 15 ps of cis ‐ 9. This should be ascribed to the more freely rotating double bond in gem ‐isomers than in cis ‐isomers. At 21 and 14 ps, the rotation was accomplished because the maximum intensity of the absorption spectra of cis ‐isomer 9and gem ‐isomer 10at the excited state was reached, respectively. It was found that the absorption maximum wavelength of the gem ‐isomer 10was shortened by 15 nm compared with that of the cis ‐isomer 9. Moreover, the area ratio of short‐wavelength band vs. long‐wavelength band was much larger for the gem ‐isomer 10than that for the cis ‐isomer 9, further corroborating that the double bond of the gem ‐isomer rotated more freely than that of the cis ‐isomer. Therefore, the gem ‐isomer showed lower fluorescence quantum yield than the cis ‐one because of the freer double bond rotation at the excited state and more nonradiative decay (see Figure 3.19).
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Handbook of Aggregation-Induced Emission, Volume 1»
Представляем Вашему вниманию похожие книги на «Handbook of Aggregation-Induced Emission, Volume 1» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Handbook of Aggregation-Induced Emission, Volume 1» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.