Handbook of Aggregation-Induced Emission, Volume 1
Здесь есть возможность читать онлайн «Handbook of Aggregation-Induced Emission, Volume 1» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Handbook of Aggregation-Induced Emission, Volume 1
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Handbook of Aggregation-Induced Emission, Volume 1: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Handbook of Aggregation-Induced Emission, Volume 1»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
A thorough introduction to the mechanistic understanding of the importance of molecular motion to aggregation-induced emission An exploration of the aggregation-induced emission mechanism at the molecular level Practical discussions of aggregation-induced emission from the restriction of double bond rotation at the excited state, and clusterization-triggered emission Perfect for academic researchers working on aggregation-induced emission, this set of volumes is also ideal for professionals and students in the fields of photophysics, photochemistry, materials science, optoelectronic materials, synthetic organic chemistry, macromolecular chemistry, polymer science, and biological sciences.
Handbook of Aggregation-Induced Emission, Volume 1 — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Handbook of Aggregation-Induced Emission, Volume 1», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
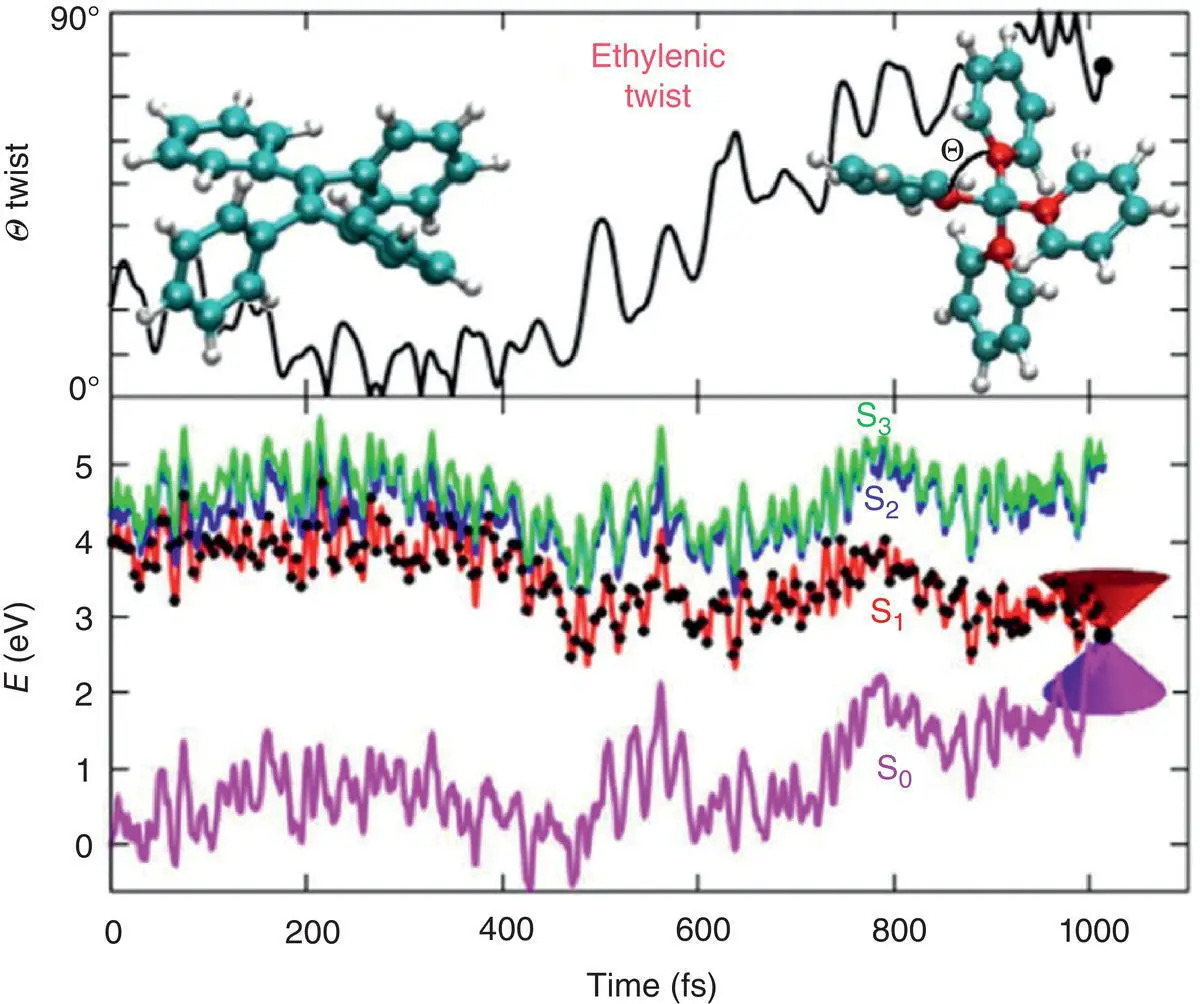
Figure 3.27 The twist angle of the double bond (upper panel) and electronic‐state potential energies (lower panel) as a function of time for two representative trajectories showing the ethylenic twist process.
Source: Reproduced with permission from Ref. [55]. Copyright 2016, Royal Society of Chemistry.
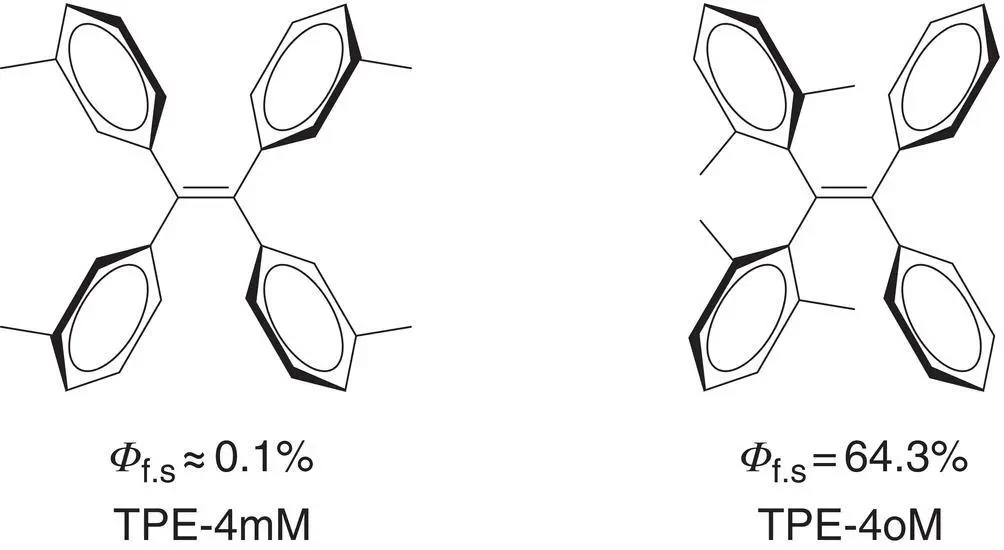
Figure 3.28 Molecular structures of TPE‐4mM and TPE‐4oM and their fluorescent quantum yields in THF ( Φ f.s) are shown below.
Thiel et al. [56] reported a calculation study of two TPE derivatives, TPE‐4mM and TPE‐4oM (see Figure 3.28right) in the isolated gas‐phase state. There is a huge difference of fluorescence quantum yields between TPE‐4mM ( Φ f= 0.1%) and TPE‐4oM ( Φ f= 64%) in solution. They combined static electronic structural calculations (TD‐DFT, CASSCF, and MS‐CASPT2) and OM2/MRCI nonadiabatic dynamics simulations to explore the nonradiative excited‐state decay mechanisms of them. The computational results showed two pairs of minimum‐energy S 1/S 0CI structures for both TPE‐4mM and TPE‐4oM. For TPE‐4mM, there was no barrier to reach the CI of photocyclization. The energy barrier for CI of the π twist was small (1.8 kcal/mol), indicating that the rotation of the double bond may also be blamed for the nonemission of TPE‐4mM in solution. But in contrast, for TPE‐4oM, the ortho‐methyl groups in TPE‐4oM effectively suppressed the rotation of both the phenyl rings and double bond. The energy barriers for the above two decay paths were non‐negligible barriers (6.2 and 8.4 kcal/mol, respectively), which prevented the nonradiative relaxation of TPE‐4oM. Consequently, the fluorescent quantum yield of TPE‐4oM was 640‐fold higher than that of TPE‐4mM in solution.
In 2018, Tang et al. [57] studied a series of TPE derivatives with varying structural rigidities and AIE properties using ultrafast spectroscopy combined with quantum computation. They found that the stretch and twist of the central double bond in TPE unit upon photoexcitation were two dominant events that caused nonradiative decay.
Figure 3.29shows the structures of TPE derivatives 18– 23in the order of increasing structural rigidity. While 18, 19, and 22showed typical AIE activity, 20displayed strong fluorescence in both solution and solid, but 21and 23have very low fluorescence quantum yields in both solutions and solids. These phenomena do not seem to match the prediction of the RIM mechanism because the fluorescence quantum yields of their solution should also gradually increase as their rigidity. However, compounds 22and 23with the most rigid structures have very low fluorescence quantum yields in solutions. In contrast, the phenyl rings of compound 20are not hinged by intramolecular cyclization, but its Φ fin solution reached an astonishing 60%. In this case, the exact mechanism that affects their fluorescent intensity in solution was worth a further study.
Firstly, they explored the geometry changes of 18– 20and 22and 23in THF solution from S 0to S 1using DFT calculation. The calculated results revealed that the absolute change of the phenyl torsion and double bond twisting in TPE derivatives decreased as the rigidity of the molecular structure increases upon excitation. In the excited state, the double bonds of TPE derivatives except for 23showed a significant extension. Compared with the emission peaks in the film, the fluorescence emission spectra in dilute solutions displayed extra peaks, which were confirmed to be the emission peaks of the photocyclization product by experiments. The above results illustrated that both double bond twisting and phenyl torsion may be responsible for the nonemission of these TPE derivatives in solutions.
Then, they further constructed the 3D potential energy surface (PES) of 18in solution (see Figure 3.30). Along the minimum energy path (MEP) of 18in the ground state, the phenyl torsion increased from 50 to 90°, but the change of the twist of double bond (<9°) and potential energy (PE) (<7 kcal/mol) was slight, indicating that the torsion of the phenyl rings dominated the ground‐state dynamics in 18. In the excited sate, the stretch and torsion of the double bond resulted in the FC* geometry changing into minimum energy geometry (S 1, minute) along the MEP. In this course, the twist of double bond was ~50°, which was accompanied by the phenyl torsion with an amplitude of less than 25°.
The ultrafast time‐resolved spectroscopy was employed to detect the geometry changes and photocyclized intermediates of 18– 23in excited state. For flexible molecules like 18, 19, and 21, the measurement demonstrated that the stretch of double bond occurred in the subpicosecond timescale (0.6–1.3 ps), and then the stretched double bond began to twist during 1.3–3.79 ps. After 3.79 ps, the photocyclization happened. For 20, due to the steric hindrance from the substituents at the o ‐position, the rotation of the double bond and photocyclization were suppressed and made the decay of emission band much longer than 18and 19. For 22or 23with a rigid structure, the formation of the photocyclized intermediate took place directly on the subpicosecond timescale so that the fluorescence was very weak.
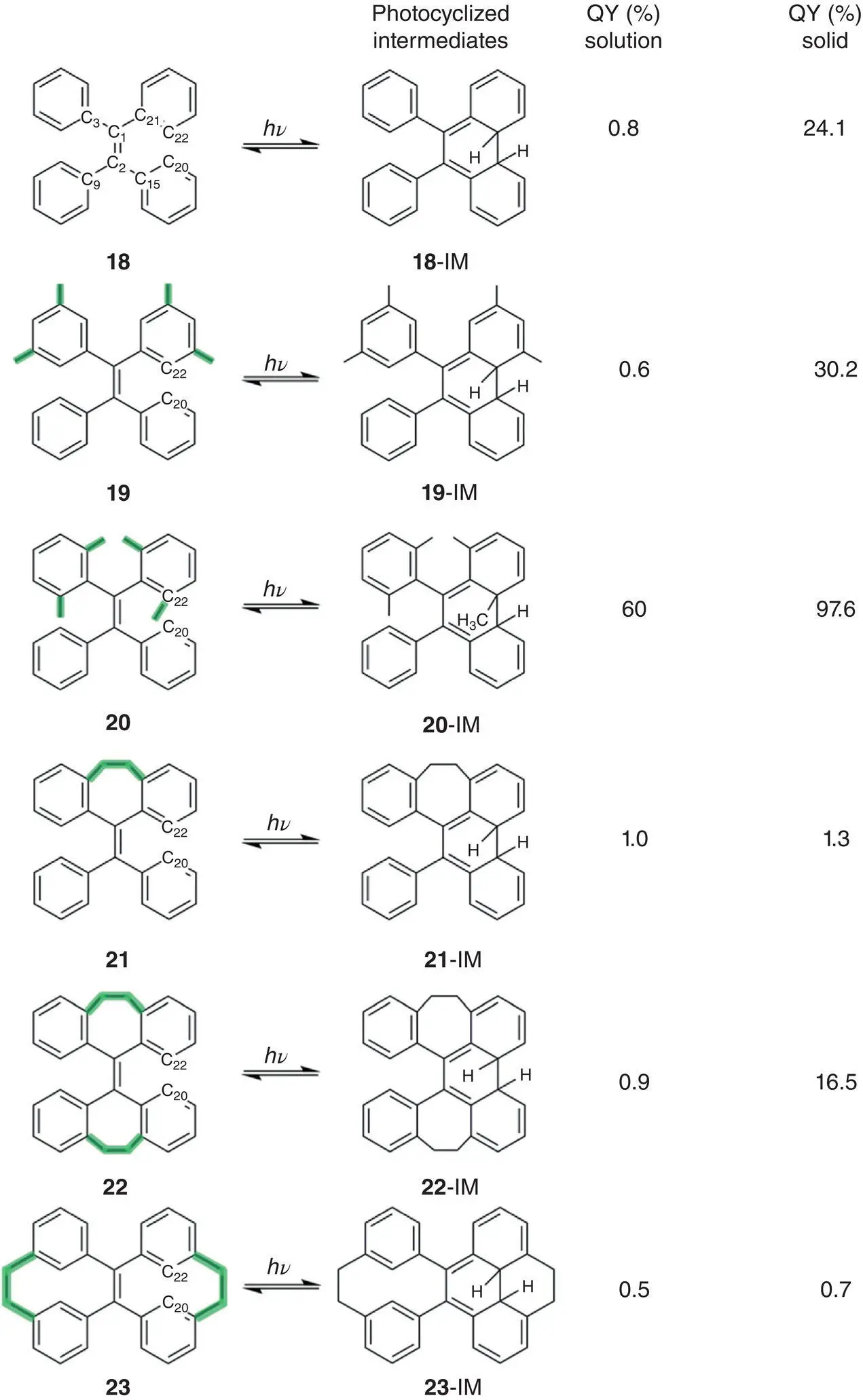
Figure 3.29 TPE derivatives 18– 23with increased structural rigidity and their transformation upon UV irradiation (QY: fluorescence quantum yield).
Source: Reproduced with permission from Ref. [57]. Copyright 2018, Royal Society of Chemistry.
There is a competitive relationship between the two processes of photocyclization and intramolecular rotation. When TPE possessed a flexible structure, the rotations of phenyl rings prohibited the photocyclization between two adjacent phenyl rings and allowed the excited double bond to rotate. But when phenyl rings are hinged by ethylene bridge, the short distance of rings promoted the ultrafast formation of the photocyclization on the subpicosecond timescale, giving no opportunity for the C═C bond to rotate. Just like molecule 20, only after these two nonradiative processes were blocked at the same time, TPE derivatives could render strong emission.
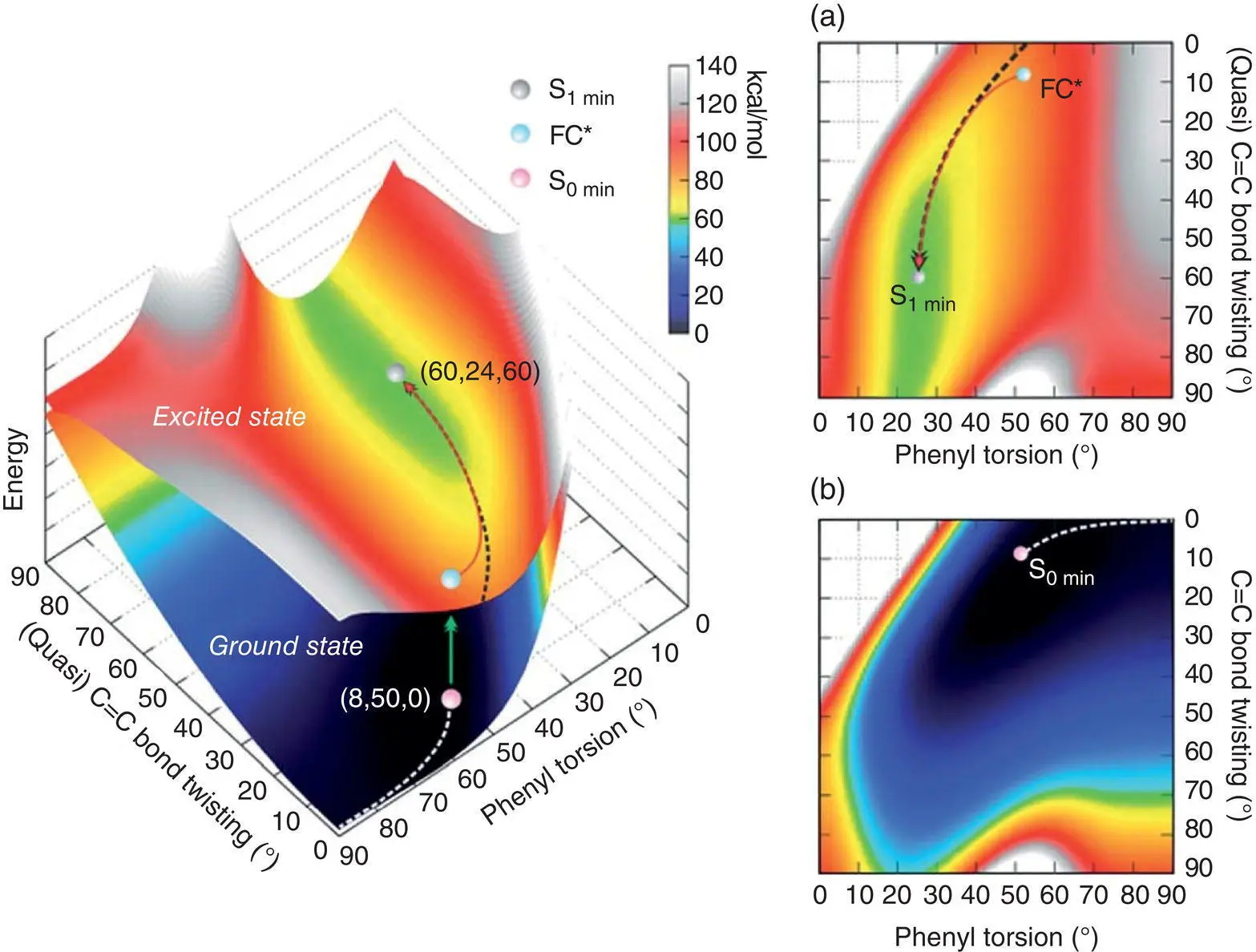
Figure 3.30 The PES of 18in the ground state and excited state as a function of the (quasi) C═C bond twisting and phenyl torsion dihedral angles. (a) Top view of the first excited‐state PES. (b) Top view of the ground‐state PES.
Source: Reproduced with permission from Ref. [57]. Copyright 2018, Royal Society of Chemistry.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Handbook of Aggregation-Induced Emission, Volume 1»
Представляем Вашему вниманию похожие книги на «Handbook of Aggregation-Induced Emission, Volume 1» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Handbook of Aggregation-Induced Emission, Volume 1» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.