James W. Brown - Principles of Microbial Diversity
Здесь есть возможность читать онлайн «James W. Brown - Principles of Microbial Diversity» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Principles of Microbial Diversity
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Principles of Microbial Diversity: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Principles of Microbial Diversity»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
provides a solid curriculum for students to explore the enormous range of biological diversity in the microbial world. Within these richly illustrated pages, author and professor James W. Brown provides a practical guide to microbial diversity from a phylogenetic perspective in which students learn to construct and interpret evolutionary trees from DNA sequences. He then offers a survey of the «tree of life» that establishes the necessary basic knowledge about the microbial world. Finally, the author draws the student's attention to the universe of microbial diversity with focused studies of the contributions that specific organisms make to the ecosystem.
Principles of Microbial Diversity
Principles of Microbial Diversity — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Principles of Microbial Diversity», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Bootstrapping
Unfortunately, it is mathematically impossible to extract confidence intervals (χ 2or p values) for nodes in a tree generated using the standard treeing methods; in other words, we cannot determine how sure we should be of the placement of each node in a tree (commonly referred to as branching order). This is not true of branch lengths, from which confidence intervals can be readily calculated. The standard method of evaluating tree branching-order reliability is called bootstrapping.
In a bootstrap analysis, the columns of a sequence alignment are randomly sampled to create a jumbled and haphazard alignment of the same length as the original. Typically 100 or 1,000 such alignments are generated from the initial alignment, and trees are generated from each. The reliability of a particular branching arrangement in a tree is judged by the frequency that the branch appears in all of the resulting trees.
The random sampling starts with the input alignment. In the standard tree generation process, the similarity matrix is generated by checking sequences against each other pairwise, tallying the similarity to each position in the alignment. Each position in the alignment is therefore counted once and only once. In a bootstrap sampling, a similarity matrix would be generated by comparing randomly selected positions in the alignment, so that some positions are compared more than once and some are not compared at all. For example, the starting alignment would be randomly sampled, ending up with a bootstrapped alignment that might look like this:
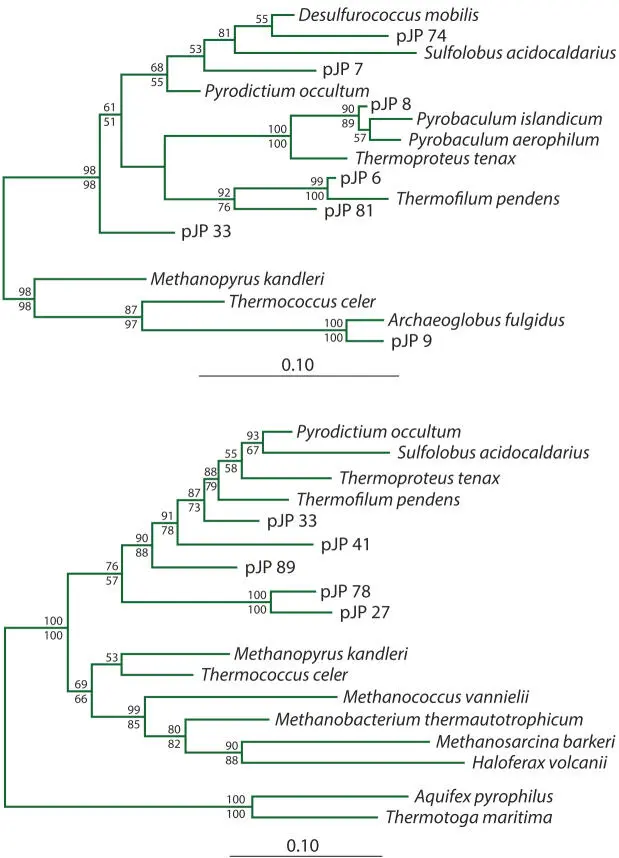
Figure 5.5Example of a published phylogenetic tree with bootstrap values included. In this case, this is a maximum-likelihood tree, with bootstrap values from both maximum-likelihood (above the branches) and maximum-parsimony (below the branches) analyses shown. (Reprinted from Barns SM, Fundyga RE, Jeffries MW, Pace NR, Proc Natl Acad Sci USA 91:1609–1613, 1994. Copyright 1994 National Academy of Sciences, USA.) doi:10.1128/9781555818517.ch5.f5.5


from which a similarity matrix and tree would be generated.
Notice that some positions in the alignment are included multiple times (9 and 2) and some are not included (4, 6, and 7). In realistically large alignments, such randomly sampled alignments yield good trees if the branching arrangements are well supported by the sequence data. Therefore, in a bootstrap analysis, a tree is generated from each of 100 to 1,000 alignments generated by random sampling of the alignment, and the number of these trees that contain each branch of the reference tree (generated in the usual way) is determined. In other words, the presence or absence of every branch in the tree is scored. The percentage of trees from a bootstrapped alignment that contain each branch in the reference tree is used to label the branches (Fig. 5.5). Often, the same type of analysis is performed using more than one method of tree construction, e.g., neighbor joining and maximum likelihood.
The evaluation of bootstrap scores is subjective, but generally branches that show up in at least 50% of trees generated from bootstrapped data sets are considered to be reliable.
Questions for thought
1 1. Compare the bootstrap values on the tree in the bootstrap section above. Which treeing algorithm seems to have generated the more robust trees, maximum likelihood or maximum parsimony? Are long or short branches more reliably predicted?
2 2. What kind of artifact would you predict from a substitution model that overestimated, instead of underestimated, the relative evolutionary distance of long branches?
3 3. During bootstrapping of an alignment, the sequences become scrambled (the residues appear out of order). Does this matter? Why or why not? (Think about it this way—would a tree come out the same if the alignment was scrambled, or not?)
4 4. In the substitution models we talked about, we focused on substitutions (obviously). How do you suppose these models deal with gaps? What about underspecified bases (e.g., R, Y, or N), or instances where sequence data are absent (i.e., in the case where only a piece of the sequence is available)?
5 5. In a parsimony analysis, a by-product of the analysis is predicted ancestral sequences. Can you think of a situation in which this might be useful?
Конец ознакомительного фрагмента.
Текст предоставлен ООО «ЛитРес».
Прочитайте эту книгу целиком, на ЛитРес.
Безопасно оплатить книгу можно банковской картой Visa, MasterCard, Maestro, со счета мобильного телефона, с платежного терминала, в салоне МТС или Связной, через PayPal, WebMoney, Яндекс.Деньги, QIWI Кошелек, бонусными картами или другим удобным Вам способом.
Интервал:
Закладка:
Похожие книги на «Principles of Microbial Diversity»
Представляем Вашему вниманию похожие книги на «Principles of Microbial Diversity» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Principles of Microbial Diversity» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.