James W. Brown - Principles of Microbial Diversity
Здесь есть возможность читать онлайн «James W. Brown - Principles of Microbial Diversity» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Principles of Microbial Diversity
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Principles of Microbial Diversity: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Principles of Microbial Diversity»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
provides a solid curriculum for students to explore the enormous range of biological diversity in the microbial world. Within these richly illustrated pages, author and professor James W. Brown provides a practical guide to microbial diversity from a phylogenetic perspective in which students learn to construct and interpret evolutionary trees from DNA sequences. He then offers a survey of the «tree of life» that establishes the necessary basic knowledge about the microbial world. Finally, the author draws the student's attention to the universe of microbial diversity with focused studies of the contributions that specific organisms make to the ecosystem.
Principles of Microbial Diversity
Principles of Microbial Diversity — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Principles of Microbial Diversity», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
2 2. What would a tree of some animals look like if constructed from globin genes where some of the sequences were alpha globins and others were beta globins? What if some of these were adult alpha or beta globins and others were juvenile or fetal globins?
3 3. What would a tree (no pun intended) of plants look like if some of the sequences (rRNAs) were accidentally taken from the chloroplast instead of the nucleus? What if all of the sequences were from the chloroplasts?
4 4. On the initial “phylum-scale” representative tree of Bacteria shown above, can you show where we might have hoped ES-2 would be?
5 5. Which properties can you predict for ES-2 based on its phylotype? Which properties can you not predict?
5
Tree Construction Complexities
The process of generating phylogenetic trees as described in chapter 4is straightforward. This is a gross simplification. Phylogenetic analysis is an entire scientific area of study, and the material that has been presented is very highly simplified. In this chapter we touch (just touch) on some of the complexities.
However, before thinking about more refined substitution models, treeing algorithms, or alternative sequences, keep one thing in mind: the most important thing by far that is needed to get a good, robust tree is to start with a good alignment. The simple and fast neighbor-joining method, using the Jukes and Cantor substitution model, usually gives perfectly usable trees if given a good small-subunit ribosomal RNA alignment to work with. Combined with bootstrapping (see below), this method is probably used more than any other for the creation of published trees.
Substitution models
In chapter 4we talked about the Jukes and Cantor method to estimate evolutionary distance from sequence similarity. This is a simple method, but there are several other more sophisticated methods. The Jukes and Cantor method and other methods for estimating evolutionary distance amount to an attempt to describe how sequences change. In other words, they are mathematical models of the process of evolution of these sequences, and they are therefore usually called “substitution models.” The choice of an appropriate substitution model is critical and often underappreciated.

Figure 5.1Two-parameter substitution models distinguish between transitions and transversions and score them differently. Each parameter is represented by an arrow. The values of these parameters can be predetermined (typically 1.0 for transversions and 0.5 for transitions) or determined by presifting the alignments to count the observed ratios of differences. doi:10.1128/9781555818517.ch5.f5.1
In the Jukes and Cantor method, any difference in two sequences is scored equivalently; for each position in a pairwise comparison, the bases are either a match or they are not. A commonly used alternative is the Kimura two-parameter model, in which transitions (purine to purine or pyrimidine to pyrimidine) and transversions (purine to pyrimidine or pyrimidine to purine) are scored differently because transitions are much more common than transversions (Fig. 5.1). These scores are based on presifting the alignment to determine the relative frequency of transitions to transversions, and these different types of changes are scored accordingly. It is even possible to have a six-parameter model, in which each type of substitution (G:A, G:C, G:U, A:U, A:C, and U:C) is scored differently (Fig. 5.2).
It is also possible to “weigh” the score of each position (column) in an alignment differently based on how conserved that position is; a difference in a conserved position is then scored as a greater difference than a difference in more variable positions. This requires alignments with many sequences so that variability at each position can be measured reliably, and so very often these are predetermined for the class of RNA being analyzed. The Weighbor algorithm used by the Ribosomal Database Project does this; the name stands for “weighted neighbor joining.” Distance matrices from protein alignments usually use a scoring table derived from the observed relative frequency with which any amino acid is substituted by another from a huge collection of aligned protein sequences, e.g., the PAM tables.
There are also different ways in which gaps can be dealt with. In most treeing algorithms, gaps are ignored; these positions are counted as neither a match nor a mismatch. This is not because they are unimportant; in fact, because insertions and deletions are less common than nucleotide substitutions, they are potentially more important than substitutions. However, it is not clear how to deal with gaps for a variety of reasons. The obvious case is where the alignment contains sequence fragments, i.e., partial sequences, instead of full-length sequences (Fig. 5.3). Partial sequences have two kinds of gaps: gaps that represent bases that are not present in that sequence (indels), and gaps that represent regions of sequence outside of the region for which the sequence is available. The algorithm cannot distinguish between these because alignments do not distinguish between different kinds of gaps.
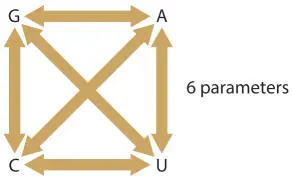
Figure 5.2A six-parameter substitution model scores each possible substitution differently. doi:10.1128/9781555818517.ch5.f5.2
It is also difficult to deal with the fact that adjacent gaps are not independent. A string of gaps probably represents the insertion or deletion of more than one base at the same time, not the one-at-a-time insertion/deletion of individual bases. For example, a five-base string of gaps most likely represents a single insertion/deletion of five nucleotides, not five independent insertions/deletions of single nucleotides. Sophisticated algorithms use a large scoring penalty for a single gap but then only a very small additional penalty for additional adjacent gaps.
In addition, it is not clear how to deal with variation at the 5′ and 3′ ends of the RNA; for example, some RNase P RNAs have the rho-independent terminator stem-loop at the end of the RNA removed while some do not (and in at least some organisms, the RNA exists in both versions). What to do with all the gaps in aligned RNAs in which this structural element is removed? And because most RNA sequences are determined from their genes, the exact ends of the encoded RNAs are most often not even known.
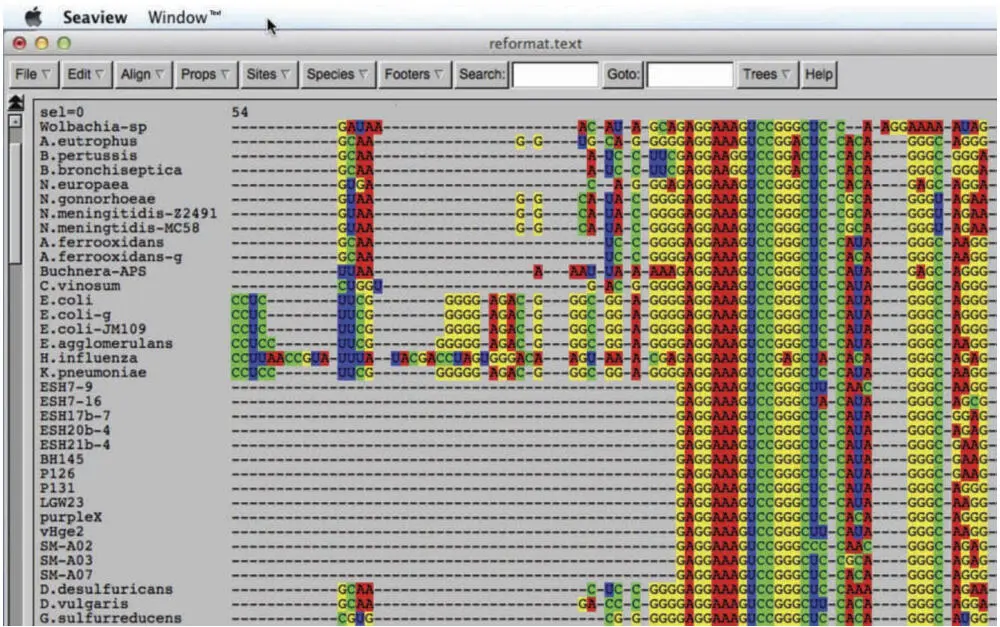
Figure 5.3An alignment showing two fundamentally different types of gaps. All of the gaps in the upper half of the sequences are indels; at least one sequence in the database has nucleotides at these positions, but these sequences do not. Some of the sequences in the bottom half of the alignment are partial sequences, i.e., sequence fragments that use gaps wherever there are no sequence data. doi:10.1128/9781555818517.ch5.f5.3
The special case of G ∙ C bias
Sometimes even rRNA sequences change adaptively—the bane of phylogenetic analysis. The most common example is the tendency of sequences to differ in G+C content, either because the genome has an unusual G+C content (i.e., there is pressure toward either G+C or A+T richness in the genome) or because the organism is a thermophile and so might prefer G=C over A=U base pairs in its RNAs. This can cause havoc in a tree. One way around this is to do a transversion analysis, which ignores transitions and only scores transversions. The common way to do this is simply to convert all of the A’s in the alignment to G’s and all U’s to C’s. Trees are generated from these alignments in the usual fashion. These trees are, of course, based on fewer data since more than half of the phylogenetic information in the alignment has been discarded, but they should be free of G+C bias artifacts.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Principles of Microbial Diversity»
Представляем Вашему вниманию похожие книги на «Principles of Microbial Diversity» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Principles of Microbial Diversity» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.