James W. Brown - Principles of Microbial Diversity
Здесь есть возможность читать онлайн «James W. Brown - Principles of Microbial Diversity» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Principles of Microbial Diversity
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Principles of Microbial Diversity: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Principles of Microbial Diversity»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
provides a solid curriculum for students to explore the enormous range of biological diversity in the microbial world. Within these richly illustrated pages, author and professor James W. Brown provides a practical guide to microbial diversity from a phylogenetic perspective in which students learn to construct and interpret evolutionary trees from DNA sequences. He then offers a survey of the «tree of life» that establishes the necessary basic knowledge about the microbial world. Finally, the author draws the student's attention to the universe of microbial diversity with focused studies of the contributions that specific organisms make to the ecosystem.
Principles of Microbial Diversity
Principles of Microbial Diversity — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Principles of Microbial Diversity», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
This works because in general it does not matter (usually) to the RNA what the bases in the helices are; what matters is that opposing bases are complementary so that they can form the helix. As a result, the secondary structure of an RNA is much more highly conserved than its sequence, because coevolution of bases that form base pairs maintains the secondary structure as the sequence changes. Variation in the length of the RNA is usually in hairpin lengthening or shortening. Therefore, it is usually possible to keep track of homologous parts of RNA structures even if the sequences are quite different.
In this type of alignment, the secondary structures of all of the RNAs are directly encoded in the alignment (Fig. 3.8). If residue n (e.g., 24 in Fig. 3.8) of any sequence pairs to residue m (e.g., 29), then so should the corresponding homologous residues in all sequences (Fig. 3.9).
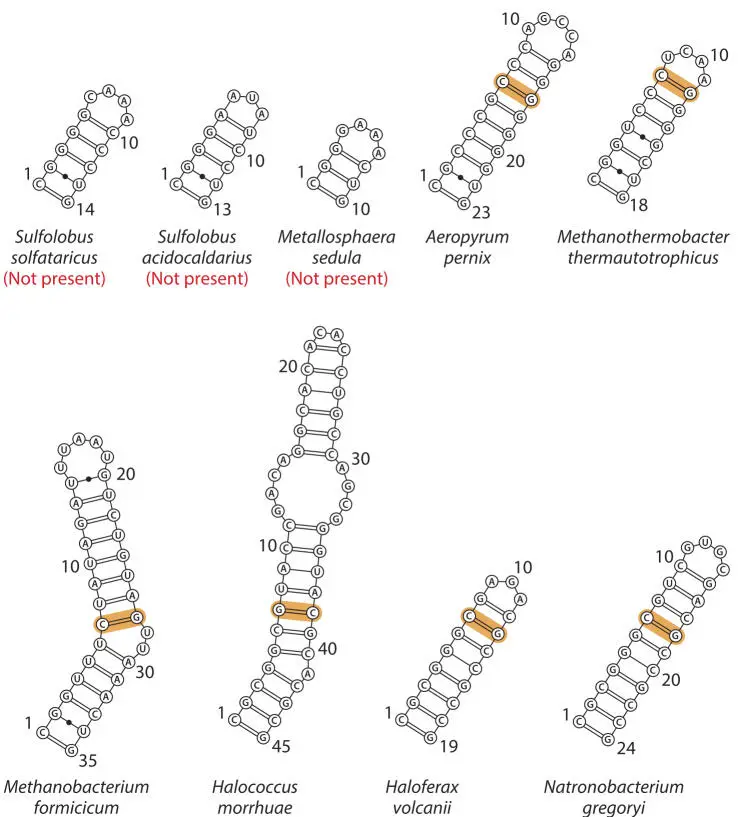
Figure 3.9RNase P RNA helix “P3” in a variety of Archaea . The base pairs corresponding to the highlighted bases in the sequence alignment in Fig. 3.8 are highlighted. P3 is present in all archaeal (and bacterial) RNase P RNAs, but both the sequence and structure of this helix are highly variable. doi:10.1128/9781555818517.ch3.f3.9
Given this type of alignment, a computer can readily compute any of the RNAs as secondary structures. Inversely, given a preexisting alignment and an RNA sequence with the same secondary structure, a computer algorithm can add this sequence correctly to the alignment. This is what infeRNAl does; it takes a sequence and tries to fold it into the correct secondary structure. If it can do so, it then threads this sequence into the alignment based on this structure.
PROBLEMS
1 1. Align the following two sequences:Now add the following sequence to this alignment:Now add the following sequence to this alignment:
2 2. Align the following sequences:
3 3. Align the following sequences:
4 4. Align the following sequences (note that these are in Fasta format, commonly used for the electronic transfer of sequence data):
5 5. Draw the secondary structures of the sequences in this alignment:
6 6. Create an alignment of the following RNA structures:
7 7. Add the following Seq V RNA structure to the preexisting alignment:
Questions for thought
1 1. What are some DNA sequences that would not be useful for phylogenetic analysis? Why?
2 2. What are some other sequences that would be useful for phylogenetic analysis, and in what situations would they be useful?
3 3. How did people get large amounts of a specific DNA for sequencing before PCR was invented?
4 4. In an episode of The X-Files (an old TV show), FBI Agent Dana Scully sequences some extraterrestrial DNA and finds “missing bands” in the sequences that she interprets to correspond to bases that are unique to aliens (not found in Earthling DNA). Why is this not technically reasonable?
5 5. Given the variation of sequences in the context of the same secondary structure, how do scientists solve these secondary structures by comparative sequence analysis?
6 6. Mutations occur one at a time. How, then, could the base pairs in a helix change without disrupting the structure of the RNA? Does this explain (at least in part) why base-pair changes that keep the purines and pyrimidines in the same positions (transitions) are more common than those that switch them (transversions)?
7 7. Although RNA three-dimensional structures are scarce, there are hundreds of protein three-dimensional structures, determined by X-ray crystallography. Can you imagine a way to use these structures, analogous to the use of RNA secondary structures, to align protein sequences more meaningfully?
4
Constructing a Phylogenetic Tree
In chapter 3, we covered the first three steps of a phylogenetic analysis, leaving the final step toward which the others build. The steps in a phylogenetic analysis are as follows:
1 1. Decide which gene and species to analyze (small-subunit ribosomal RNA [SSU rRNA])
2 2. Determine the gene sequences (polymerase chain reaction [PCR] and DNA sequencing, database “mining”)
3 3. Identify homologous residues (sequence alignment)
4 4. Perform the phylogenetic analysis
The most common type of phylogenetic analysis is tree construction. A tree is nothing more than a graph representing the similarity relationships between the sequences in an alignment. This is why we’ll be going through this process in such detail, to show that tree construction is not rocket science but involves straightforward mathematical transformations of sequence data.
There are several methods for building trees. In this chapter, we cover the neighbor-joining method in some detail as an example, because it is conceptually straightforward and commonly used. In the next chapter, we briefly cover some other approaches.
Tree construction: the neighbor-joining method
Tree construction starts with an alignment. Neighbor joining is a distance matrix method, meaning that the alignment is first reduced to a table of evolutionary distances, a distance matrix. The distance matrix cannot be generated directly from the alignment, however, because actual evolutionary distance cannot be directly measured. Instead, the alignment is reduced to a table of observed (measurable) similarity, the similarity matrix. The distance matrix is calculated from the similarity matrix, and then the tree is generated from the distance matrix.
Generating a similarity matrix
The similarity matrix is just a table of fractional similarities, for example, in this alignment of six sequences with 20 positions.
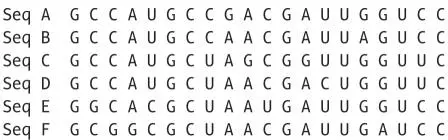
Just count the fraction of identical bases in every pair of sequences in the alignment.
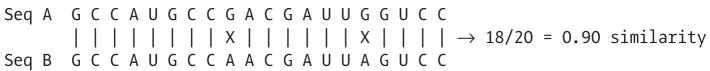
The similarity values for all pairs of sequences are calculated in the same way and assembled into a table:
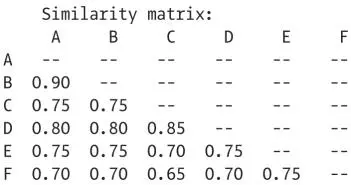
In this example, sequences A and B are 0.90 (90%) similar, A and C are 0.75 similar, B and C are 0.75 similar, and so forth. Note that values on the diagonal (A:A, B:B, …) do not need to be calculated; they are always 1. Likewise, there is no reason to calculate both above and below the diagonal; the value for X:Y is the same as that for Y:X, so the second calculation would be redundant.
Converting a similarity matrix into an evolutionary distance matrix
Next is the estimation of evolutionary distances from their sequence similarity. You might think that the distance would just be 1 − similarity (i.e., “difference”), and you would be right except that the number of differences you count between any two sequences misses some of the changes that probably have occurred between them. More than one evolutionary change at a single position (e.g., A to G to U, or A to G in one sequence and the same A to U in another) counts as only one difference between the two sequences, and in the case of reversion or convergence it counts as no change at all (e.g., A to G to A, or A to G in one organism and the same A to G in another). As a result, the observed similarity between two sequences underestimates the evolutionary distance that separates them.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Principles of Microbial Diversity»
Представляем Вашему вниманию похожие книги на «Principles of Microbial Diversity» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Principles of Microbial Diversity» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.