James W. Brown - Principles of Microbial Diversity
Здесь есть возможность читать онлайн «James W. Brown - Principles of Microbial Diversity» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Principles of Microbial Diversity
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Principles of Microbial Diversity: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Principles of Microbial Diversity»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
provides a solid curriculum for students to explore the enormous range of biological diversity in the microbial world. Within these richly illustrated pages, author and professor James W. Brown provides a practical guide to microbial diversity from a phylogenetic perspective in which students learn to construct and interpret evolutionary trees from DNA sequences. He then offers a survey of the «tree of life» that establishes the necessary basic knowledge about the microbial world. Finally, the author draws the student's attention to the universe of microbial diversity with focused studies of the contributions that specific organisms make to the ecosystem.
Principles of Microbial Diversity
Principles of Microbial Diversity — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Principles of Microbial Diversity», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
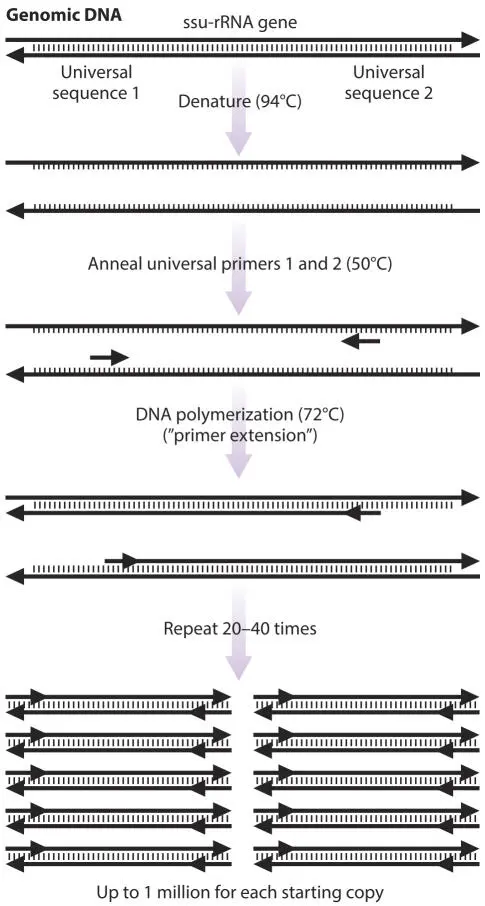
Figure 3.3The polymerase chain reaction (PCR). doi:10.1128/9781555818517.ch3.f3.3
The PCR product DNA is then sequenced (i.e., its nucleotide sequence is determined), often using the same oligonucleotide primers that were used in the PCR. Sequencing involves denaturing the DNA, annealing an oligonucleotide primer, and extending from this primer with DNA polymerase in the presence of deoxynucleoside triphosphates (dNTPs) and small amounts of chain terminator dideoxynucleotides (analogs of dNTPs from which DNA polymerase cannot continue extending) (Fig. 3.4). Usually this process is carried out by a commercial service rather than in a research lab. A fluorometer at the bottom of the gel or end of the capillary detects the termination dyes as they run past. The connected computer collects this data and reads the sequence from the pattern of peaks (Fig. 3.5).
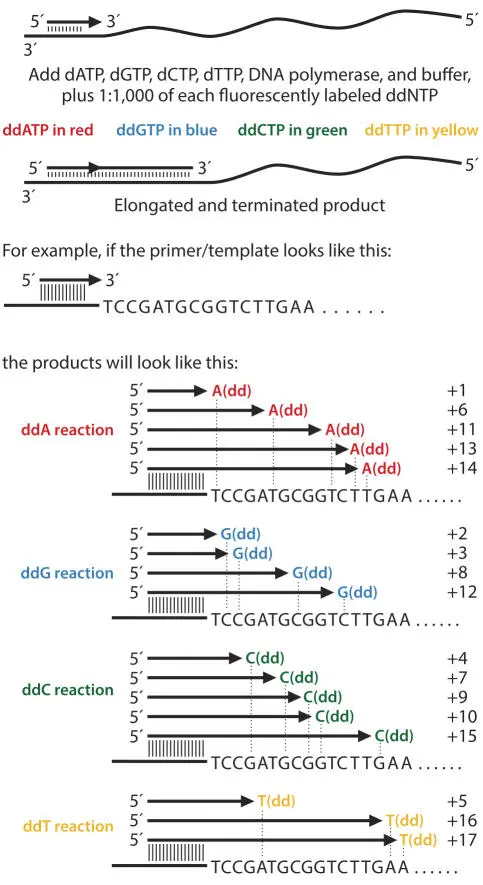
Figure 3.4Chain termination sequencing. doi:10.1128/9781555818517.ch3.f3.4
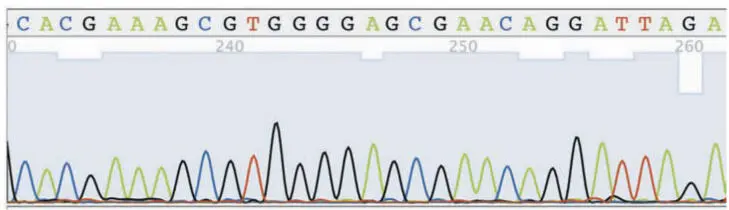
Figure 3.5Example sequence data from a DNA sequencing reaction. doi:10.1128/9781555818517.ch3.f3.5
Each reaction typically yields 500 to 1,000 bases of reliable sequence data, so it is usually necessary to use several primers spaced along the length of the molecule to get the complete sequence of an rRNA gene. It is also usually expected that both strands of the DNA will be sequenced for confirmation.
Assembling sequences in a multiple-sequence alignment
The raw material used by a phylogenetic tree-generating program is an alignment (Fig. 3.6). A sequence alignment is a two-dimensional matrix of multiple sequences. Each sequence is in a line (row) of the matrix. Each position (column) in an alignment contains homologous (corresponding) residues of each sequence. Gaps (usually shown as dashes) are added where needed to maintain the alignment; these gaps represent the absence of bases in the sequence that are present in some other sequence(s) in the alignment.
Alignment based on conserved sequence
Most alignments are generated by using computer programs that align sequences from algorithms (e.g., CLUSTAL) that attempt to maximize the similarity (measured in a variety of ways) of all of the sequences. Where the sequences in an alignment are very similar, this approach can generate very good alignments. This is especially true for protein-encoding sequences, with 20 possible amino acids and good scoring matrices to count how similar or different any two amino acids are from each other. This is less true for DNA or RNA sequences, with only four possible bases and where similarity between pairs of bases is less meaningful in the context of the encoded macromolecule.
Very often, however, RNA alignments are either created by hand or at least adjusted manually. Sequences must be fairly similar in sequence and length to be readily aligned by eye or even by computer alignment programs. However, most of the length of SSU rRNAs is highly conserved and can with experience be manually aligned without much trouble.
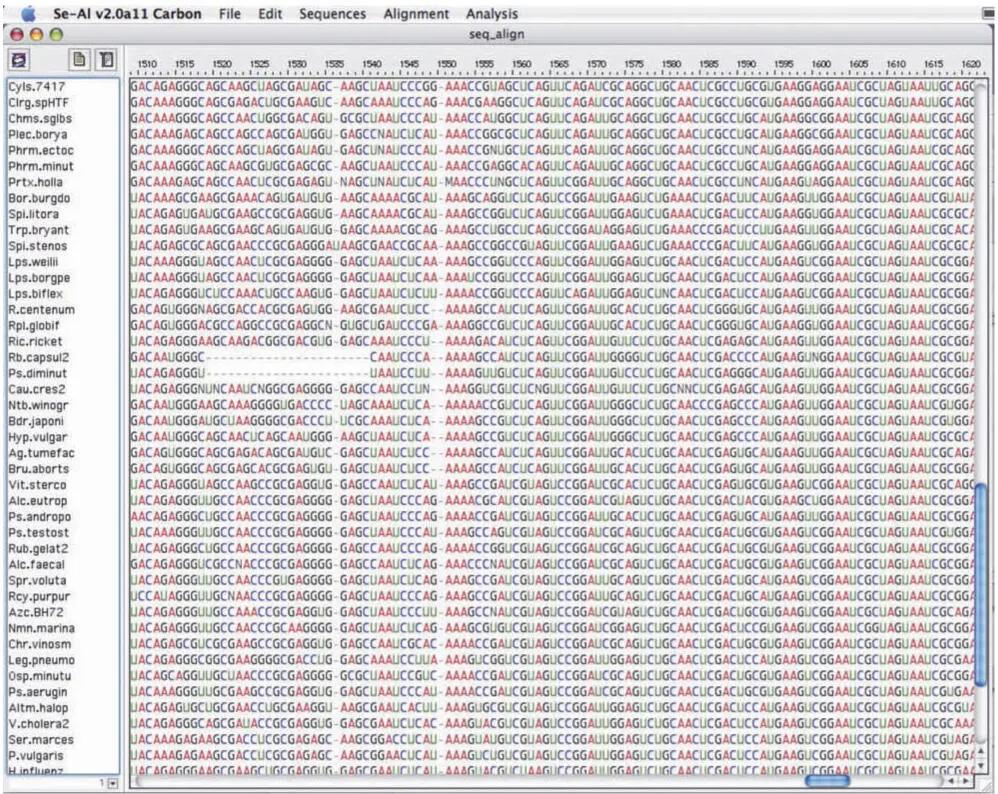
Figure 3.6A small window into an alignment of SSU rRNA sequences. doi:10.1128/9781555818517.ch3.f3.6
Some of the tricks to aligning sequences by hand are the following.
Sequences are often aligned sequentially; start by aligning the two most similar sequences, then add sequences to the alignment one at a time, starting with the sequences most similar to those already aligned and finishing with the most distantly related sequences. Likewise, if you are adding a single sequence to an existing alignment, start by identifying the most similar sequence in the alignment and use that sequence as a guide.
Alternatively, you can identify conserved blocks of sequence in all of the sequences and align these. You have now broken the alignment problem into smaller, easier chunks. Add gaps as needed to align the space between prealigned chunks according to the criteria below.
Start by finding patches of very similar sequences and align these, then work out in both directions from these, adding gaps sparingly when needed. Everything after this is about rearranging (and potentially adding or removing) these gaps.
Where there are sequence differences, slide the gaps around to keep purines (G, A) aligned with purines, and pyrimidines (C, U/T) aligned with pyrimidines.
Try also to keep differences together in variable sequence positions, and align gaps together in columns wherever possible. A single gap of two positions is a lot better than two separate gaps of one position each.
Try to keep what look like conserved positions (columns) conserved, and all things being equal, put differences into positions already known to be variable.
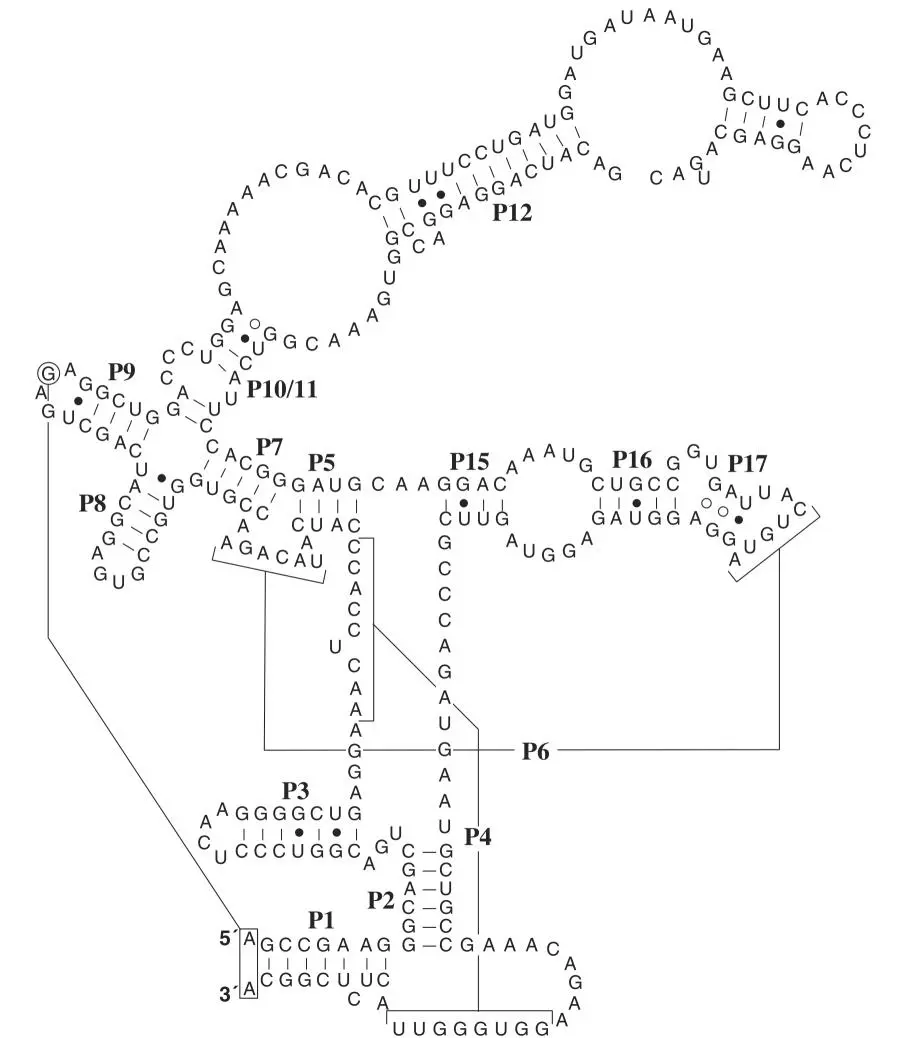
Figure 3.7AComparison of two RNase P RNAs with very different sequences and very similar secondary structures. RNase P RNAs are the catalytic subunits, associated with one or more accessory proteins, that remove the 5′ leaders from tRNA and other RNA precursors. (Adapted from Harris JK, Haas ES, Williams D, Frank DN, Brown JW, RNA 7:220–232, 2001, with permission.) doi:10.1128/9781555818517.ch3.f3.7A
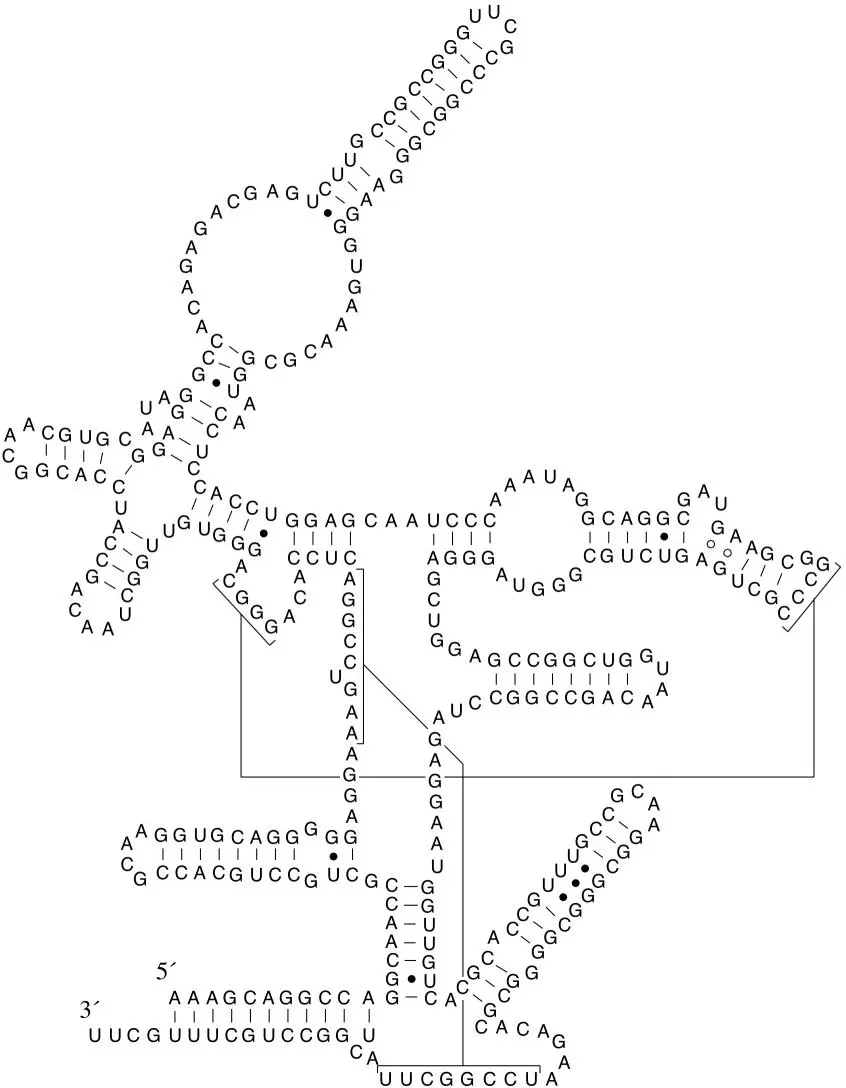
Figure 3.7Bdoi:10.1128/9781555818517.ch3.f3.7B
Alignment based on conserved structure
In the case of RNAs, however, advanced alignment algorithms (e.g., infeRNAl) can use the secondary structures of the RNAs to align sequences. The ability to use well-defined secondary structures to identify homologous residues (i.e., to align sequences) is one of the key advantages of RNA over protein for phylogenetic analysis. In other words, you can use the secondary structure of the RNA to identify homologous parts of the RNA, rather than relying only on sequence similarity (Fig. 3.7).

Figure 3.8An RNA alignment based on secondary structure. If residue n (e.g., 24, highlighted) of any sequence pairs to residue m (e.g., 29, also highlighted), then so should the corresponding homologous residues in all sequences. This is an RNA alignment based on secondary structure: stem-loop P3 of RNase P RNA. In this example, the first six rows are not sequences, they are annotations. The first three are just a reference numbering; in this case, the Methanothermobacter thermautotrophicus (Mthermo) sequence is the reference sequence. The row marked “helices” indicates the secondary structure: the 5′ strand of P3 followed by the loop and then the 3′ strand. Each base pair in this stem-loop is indicated by matching right- and left-facing parentheses in the following row and is labeled alphabetically (for human readability) in the subsequent row. doi:10.1128/9781555818517.ch3.f3.8
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Principles of Microbial Diversity»
Представляем Вашему вниманию похожие книги на «Principles of Microbial Diversity» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Principles of Microbial Diversity» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.