James W. Brown - Principles of Microbial Diversity
Здесь есть возможность читать онлайн «James W. Brown - Principles of Microbial Diversity» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Principles of Microbial Diversity
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Principles of Microbial Diversity: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Principles of Microbial Diversity»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
provides a solid curriculum for students to explore the enormous range of biological diversity in the microbial world. Within these richly illustrated pages, author and professor James W. Brown provides a practical guide to microbial diversity from a phylogenetic perspective in which students learn to construct and interpret evolutionary trees from DNA sequences. He then offers a survey of the «tree of life» that establishes the necessary basic knowledge about the microbial world. Finally, the author draws the student's attention to the universe of microbial diversity with focused studies of the contributions that specific organisms make to the ecosystem.
Principles of Microbial Diversity
Principles of Microbial Diversity — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Principles of Microbial Diversity», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Clock-like behavior depends mostly on functional constancy of the sequence; a change in the function (or functional properties) leads to large, selected (and therefore nonrandom) sequence change, i.e., adaptation. Clock-like behavior also depends on the sequence being long enough to provide statistically significant information and being made up of a large number of independently evolving “bits” so that random changes in one part of the sequence do not influence changes in other parts of the sequence. The sequence must also have an appropriate amount of sequence variation; too little variation does not provide enough difference to be statistically meaningful, whereas too much makes alignment difficult or impossible and decreases the reliability of the treeing algorithm (see chapters 4and 5on evolutionary models). Nonfunctional sequences (e.g., some introns) usually change too fast for analysis except of the very closest of relatives.
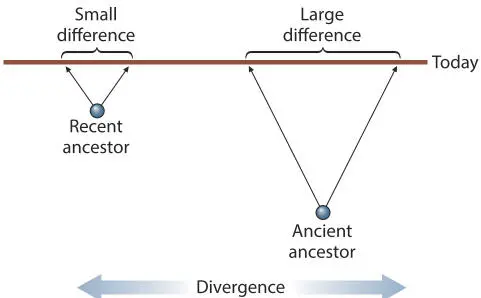
Figure 3.1Clock-like behavior. The extent of sequence divergence between a pair of specific sequences should be a measure of how long ago they separated. doi:10.1128/9781555818517.ch3.f3.1
Phylogenetic range
In order to be useful for a phylogenetic analysis, a sequence must be present and identifiable in all of the organisms to be analyzed and must exhibit clock-like behavior within this range. Watch out for gene families, because each member of the family is probably specialized for a slightly different function and it is often difficult to identify the correct ortholog or confirm that it really does have the same function.
Absence of horizontal transfer
Absence of horizontal transfer means that the gene must be acquired only by inheritance from parent to offspring, not by transfer from one organism to another except by descent. Examples of frequently horizontally transferred genes are those encoding antibiotic resistance, but any gene has the potential to be transferred horizontally. You can still generate a tree with sequences that have been horizontally transferred, but if the sequence is otherwise a good molecular clock, the resulting perfectly valid tree will reflect the phylogenetic relationships between the sequences but not the organisms that carry these sequences.
Availability of sequence information
It is of great pragmatic importance to choose a sequence, whenever possible, for which a great deal of the sequence data required is already available and annotated and perhaps already aligned. If you are interested in the phylogenetic placement of organism X , it is better if you do not have to obtain or identify the sequence data yourself for a large number of organisms to which it might (or might not) be related.
The standard: small-subunit ribosomal RNA
In most cases, the best molecular clock for phylogenetic analysis is the small-subunit ribosomal RNA (SSU rRNA) (Fig. 3.2). This sequence is always the best starting point; only after you know where your organism resides in an SSU rRNA phylogenetic tree can you decide what other sequences might provide additional information (see chapter 6for alternatives).
The SSU rRNA is so often the best sequence of choice for the following reasons.
It is present in all living cells.
It has the same function in all cells.
It comprises 1,500 to 2,000 residues—large enough to be statistically useful but not too large to be onerous to sequence.
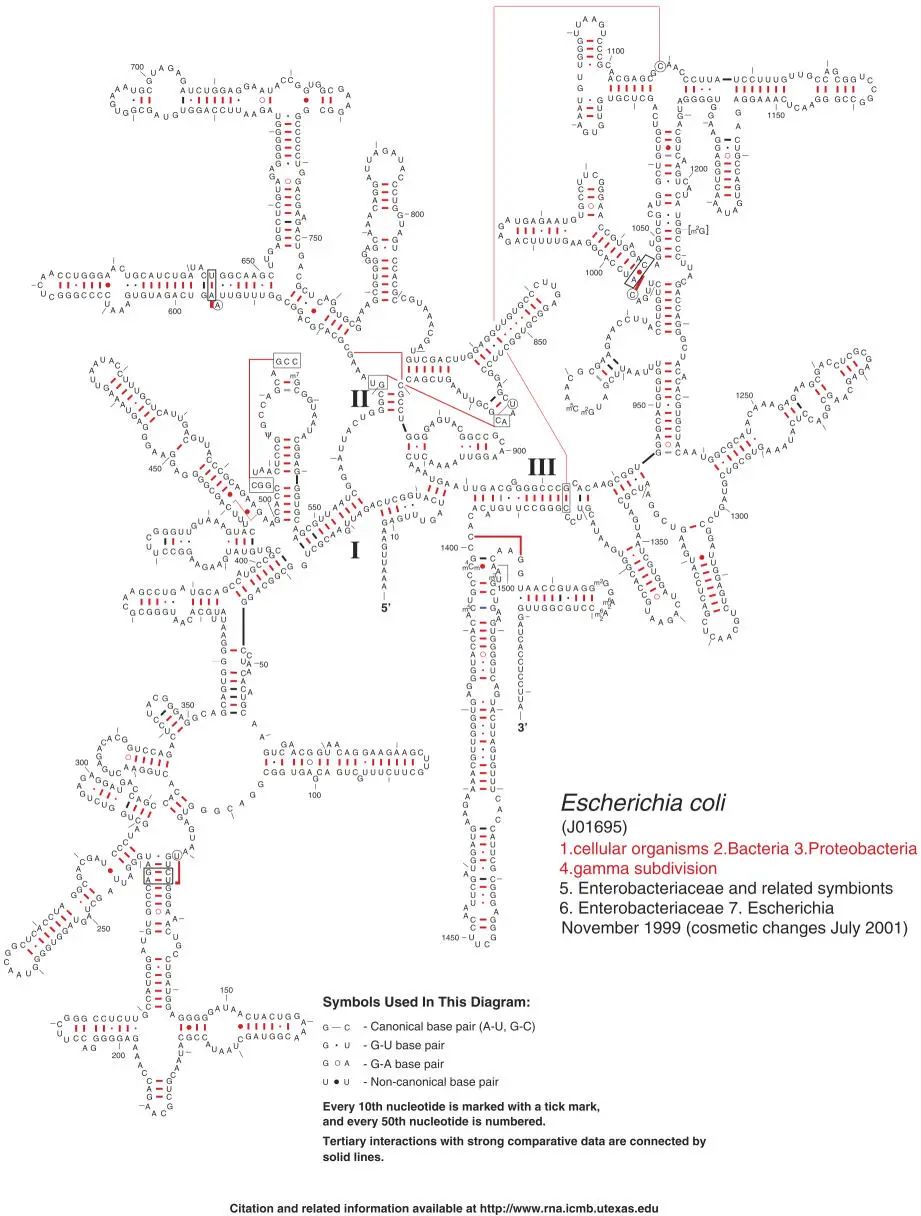
Figure 3.2The Escherichia coli SSU rRNA secondary structure. (Courtesy of Robin Gutell. Adapted from Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Müller KM, Pande N, Shang Z, Yu N, Gutell RR, BMC Bioinformatics 3:2, 2002. doi:10.1186/1471-2105-3-2) doi:10.1128/9781555818517.ch1.f1.11B
It is made up of ca. 50 independently evolving helices and ca. 500 independently evolving base pairs.
It is conserved highly enough in sequence and structure to be easily and accurately aligned.
It contains both rapidly and slowly evolving regions—the rapidly evolving regions are useful for determining close relationships, whereas the slowly evolving regions are useful for determining distant relationships.
Horizontal transfer of rRNA genes is exceedingly rare (most genes of the central information-processing pathways of the cell are also resistant to horizontal transfer).
Huge data sets of sequences, alignments, and analysis tools are available.
Deciding which organisms to include
Usually, deciding which organisms to include is part of the treeing process rather than something done in advance. As is explained in chapter 4, most often you start out by generating a tree with representatives from a wide range of organisms scattered around the tree in order to identify what kind of organism it is in very general terms, and then you replace most of these disparate representatives with representatives that you now know are likely to be closely related. The resulting tree gives you more specific information about the group to which your organism belongs, which can be used again to choose even closer relatives, and so on until you are satisfied with the representation of the tree. For example, if you have a new organism to identify, an initial tree containing one or two representatives from each bacterial phylum might show you that your organism is a member of the Firmicutes . With this information in hand, a second tree populated by representatives of each order and family of Firmicutes might show you that you might have a member of the family Veillonellaceae . From there, you could populate a final tree with most of the species in this family.
Obtaining the required sequence data
Sequences for a phylogenetic analysis usually come from two sources: electronic databases and your own experimental results. Ideally, all of the sequence data needed can be obtained from databases, or at least all of the data needed except that of the specific organism(s) of interest. However, you might find that some other sequences you need for comparison are unavailable; if this is the case, you may need to obtain them yourself experimentally.
Databases
For most sequences commonly used for phylogenetic analysis, there are specialized databases of pre-aligned sequences. There are several databases of SSU rRNAs and their alignments: the Ribosomal Database Project is a prime example and is specialized for phylogenetic analysis. As of this writing, the Ribosomal Database Project contains 1.3 million aligned SSU rRNA sequences and a suite of software tools to access these data, including a taxonomic browser that can be used to collect any desired aligned sequences for further analysis. Databases of this type are usually the best starting point from which to collect an initial data set.
Very often, however, there is additional useful information that is not yet available in these specialized databases. BLAST searches of general sequences databases (e.g., GenBank), most often through the National Center for Biotechnology Information website, often identify additional useful sequences.
Obtaining sequences experimentally
The commonly used method to obtain DNA for sequence analysis is the polymerase chain reaction (PCR). PCR amplifies genes exponentially—a single molecule of a gene, embedded in the rest of the genomic DNA, is specifically amplified to up to a million molecules in just a couple of hours. In a PCR, three steps (denaturation, primer annealing, and DNA polymerization) are cycled over and over, each time doubling the amount of the specific DNA fragment (Fig. 3.3).
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Principles of Microbial Diversity»
Представляем Вашему вниманию похожие книги на «Principles of Microbial Diversity» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Principles of Microbial Diversity» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.