Spectroscopy for Materials Characterization
Здесь есть возможность читать онлайн «Spectroscopy for Materials Characterization» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Spectroscopy for Materials Characterization
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Spectroscopy for Materials Characterization: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Spectroscopy for Materials Characterization»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Learn foundational and advanced spectroscopy techniques from leading researchers in physics, chemistry, surface science, and nanoscience Spectroscopy for Materials Characterization,
Simonpietro Agnello
Spectroscopy for Materials Characterization
Spectroscopy for Materials Characterization — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Spectroscopy for Materials Characterization», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
It is known [68] that photoexcitation within the singlet 1MLCT state is typically followed by relaxation into a long‐lived triplet 3MLCT state, through an intersystem crossing (ISC) process, which can be surprisingly fast, well in the sub‐picosecond range. Because of such an extremely short 1MLCT fluorescence lifetime, its quantum yield is so small to make it practically invisible in steady state measurements. For the same reason, a direct observation of ISC in real time is very challenging, but can be achieved by FLUC, which allows to directly detect 1MLCT fluorescence and follow its dynamics as a function of time.
An example of this is reported by Cannizzo et al. [34]. The authors of this paper investigated the dynamics of an archetypal metal complex ([Ru( bpy ) 3] 2+) by ultrafast fluorescence upconversion. In particular, they reported for the first time a FLUC experiment where the detection covered a broad wavelength range (440–690 nm) and, simultaneously, a time resolution of ≈110 fs. Photoexciting the metal complex with a very short excitation pulse at 400 nm allows them to follow the dynamics from the earliest stage, thus capturing the whole relaxation of the system.
The signal observed by FLUC [34] can be described as an emission band, peaking at 19 230 cm −1, which is attributed to 1MLCT emission and grows together with the excitation pulse. This band then decays within 200 fs while a weaker band appears at 16 500 cm −1. The latter corresponds to the steady state emission from the triplet state 3MLCT.
The detailed analysis of the temporal kinetics demonstrates that the bluest 1MLCT band rises and decays during the duration of the excitation pulse, and that also the rise of the redder band occurs during the pulse duration, suggesting that the population of the 3MLCT state is driven by an extremely fast ISC which lasts much less than the experimental time resolution. Deconvolution of the instrumental response function suggests an ISC timescale smaller than 30 fs, which implies in turn an extremely efficient spin–orbit coupling for this complex.
The shape of 1MLCT emission at zero time involves a large excitation–emission Stokes shift immediately after excitation. Analysis of the bandshape initially led to hypothesize that this transient 1MLCT emission is vibrationally “hot,” because 400 nm pump pulses selectively excite the v = 2 level of the Franck–Condon progression in absorption [34]. However, this issue was further addressed by later studies by the same group, which revealed instead that the transient 1MLCT fluorescence is vibrationally relaxed on ultrashort timescales [69], possibly even faster than the ISC process.
3.6.2 Relaxation of Excited Charge Carriers and Excitons in Semiconductor Nanoparticles
Ultrafast transient absorption experiments help to investigate, as already said, the sub‐picosecond and picosecond dynamics occurring in different materials after photoexcitation. As a further example, here we report some ultrafast TA studies conducted by Klimov's group ([27]) on CdSe nanocrystals aiming to investigate the fast carrier dynamics of electron–hole pairs and the effects produced by very intense pump excitation, capable of generating multiple excitations in the same nanocrystal.
The experiment consisted in the photoexcitation of CdSe colloidal NCs excited with 100 fs pulses peaking at 400 nm (3.1 eV, that is higher than CdSe bandgap) and in the consequent probing of the absorption changes in the visible‐IR range. The size of the studied nanocrystals is small enough to fall in the strong confinement regime (diameter smaller than Bohr radius), characterized by an energy spacing between the lowest electron states larger than both the exciton binding energy and the longitudinal optical phonon. The results obtained by TA provided a comprehensive picture of the dynamics of photoexcited charge carriers from the very first moment after photoexcitation.
Figure 3.8a reports typical TA spectra observed in the system at different delays from photoexcitation. The features appearing in the TA spectra can be given a definite identity by a direct comparison with features present in the steady state spectrum. For example, the lowest‐energy feature B1 clearly visible in both the TA and steady state spectra is associated to e–h pairs formed by an electron in the 1S state and a hole in the 1S 3/2state. The spectra at relatively long times (ps) are mostly explained by the bleach of several features of the absorption spectrum, due to the reduced absorption after one (at low pump intensities) or more e–h couples have been generated by photoexcitation. These bleaching signals, also named state filling effects, appear as positive signals in Figure 3.8a because the TA is reported with inverted sign (−Δ α ). Besides state filling effects, a peculiar derivative‐like shape, composed by features B1 and A1, shows up in the TA at short times. This feature is attributed to bi‐exciton interactions, that are the Coulombic interactions between the e–h pairs generated by the pump and the probe. On increasing pump–probe time delay, carriers relax from initially high excited to low‐energy states, the coulombic contributions diminish and the state filling contributions dominate the spectra. Therefore, the spectral evolution in Figure 3.8a provides direct information on the timescales of the carrier energy cascade in semiconductor NCs.
Beyond the timescales of carrier dynamics, TA signals collected at different pump powers ( Figure 3.8b) provide some useful additional information. When the pumping intensity is low enough, the photoexcited average NC population can be estimated to be lower than 1, that is less than one e–h pair generated on average per NC. However, if the power is increased in order to have average NC population greater than 1, new decay components show up in the temporal kinetics of the TA signals, as clearly visible in Figure 3.8b. These components are due to the activation of new non‐radiative depopulation channels in the sub‐nanosecond regime, due to the Auger recombination events which become possible whenever multiple e–h pairs coexist in the same quantum dot. Interestingly, the Auger lifetimes turn out to be quantized, because the number of generated e–h pairs can only be discrete, yielding a progression of decay lifetimes which depend on pump power ( Figure 3.8b). Therefore, the carrier dynamics result to be faster if the power and, consequently, the number of e–h pairs per NC increase.
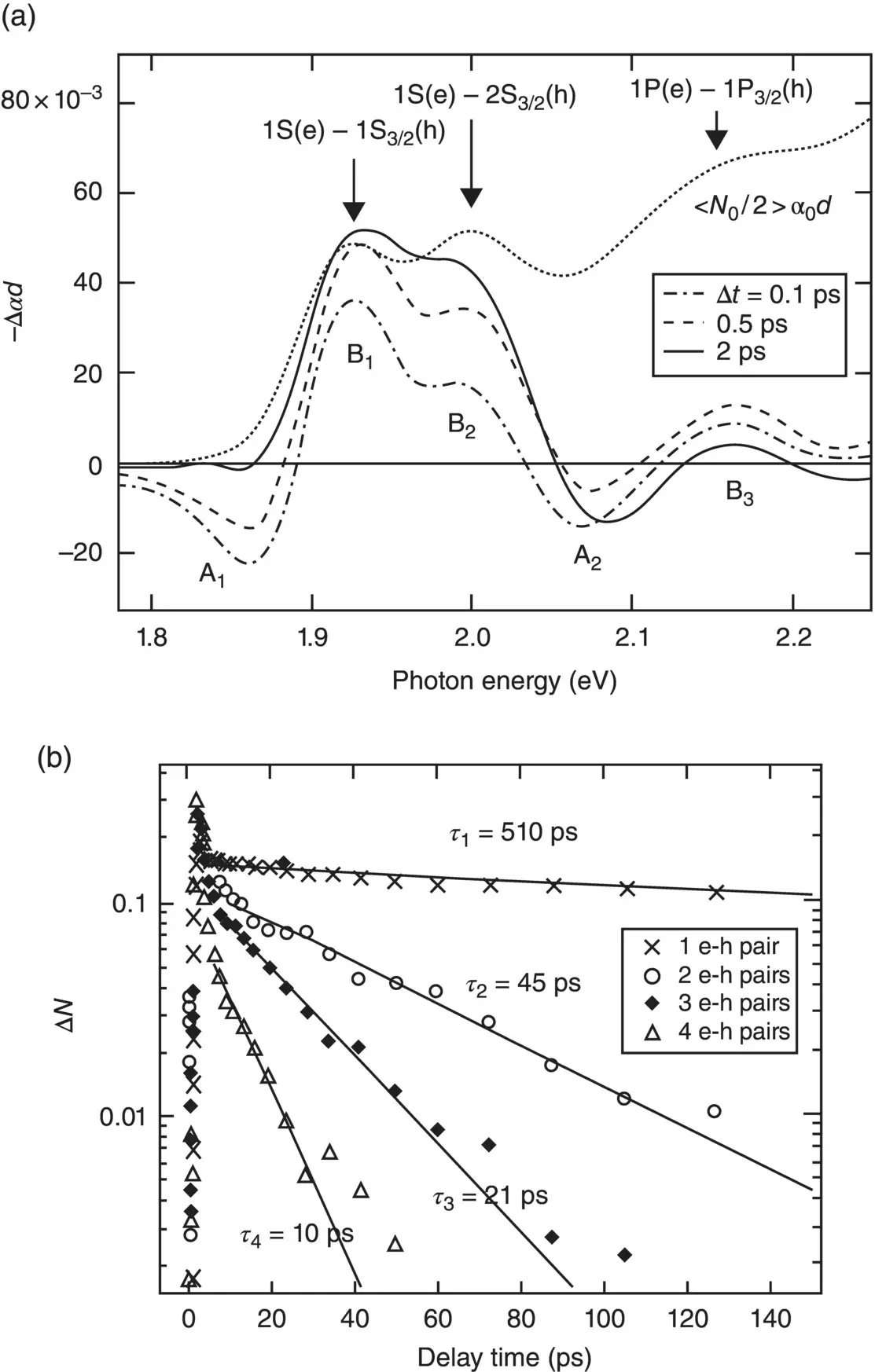
Figure 3.8(a) TA spectra of CdSe NCs recorded at 0.1, 0.5, and 2 ps compared with the steady state absorption. The features A iare associated to bi‐exciton Coulombic interactions decaying in time, while features B iare due to depopulation of the ground state. (b) TA kinetics of the 1‐, 2‐, 3‐, and 4‐ e–h couples fit to a single‐exponential decay.
Source: Adapted with permission from Klimov [27]. Copyright (2000) American Chemical Society.
3.6.3 Ultrafast Relaxation Dynamics of Carbon‐based Nanomaterials
Besides the investigation of ultrafast relaxations within a given physical system, such as a molecule, solid, or nanomaterial, femtosecond techniques allow to investigate the interactions of materials with the external environment and with external agents, often characterized by very fast photoreactions. As an example, we report an ultrafast study of the interaction of metal ions with carbon nanodots (CDs), a recently discovered family of fluorescent carbon‐based nanoparticles [29]. In particular, the aim of the work was to investigate the changes of the optical properties of CDs induced by metal ions in solution, demonstrating the occurrence of an emission quenching and explaining the underlying mechanism.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Spectroscopy for Materials Characterization»
Представляем Вашему вниманию похожие книги на «Spectroscopy for Materials Characterization» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Spectroscopy for Materials Characterization» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.