Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
Здесь есть возможность читать онлайн «Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
The first book dedicated to the emerging field of physiologically based pharmacokinetic modeling (PBPK) Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulations: Principles, Methods, and Applications in the Pharma Industry
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry, Second Edition
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
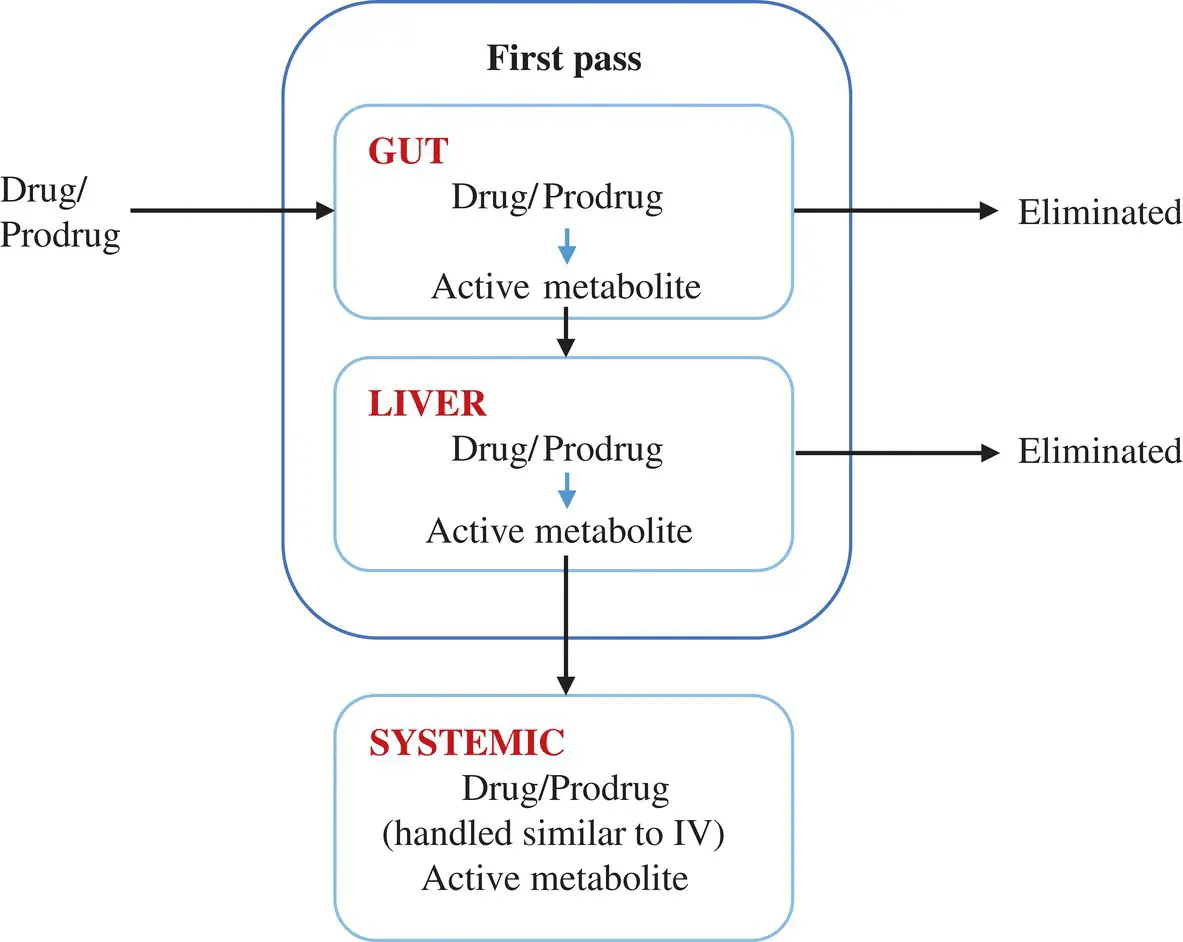
Figure 1.10. An orally administered drug or a prodrug may undergo first‐pass metabolism both in the gut and liver resulting in the formation of active metabolite. Both drug and metabolite may be eliminated in each of these organs. For simplicity, the active metabolite formed in the two organs are generally assumed to be completely available in systemic circulation without any elimination in the organ of origin. The fraction of drug bioavailable after first‐pass extraction in systemic circulation is handled similar to IV. Therefore, the amount of metabolite at a given time in systemic circulation is the sum of the amounts of active metabolite from first‐pass and from the biotransformation of the bioavailable fraction of parent drug.
TABLE 1.3. Impact of changes in biological parameters on pharmacokinetic properties.
| PK property | Change in biological parameter | Causes | Impact on PK |
|---|---|---|---|
| Absorption | Decrease in small intestinal surface area Reduced gastric emptying rate Increase in gastric pH | Disease or age Fed state; type of food Disease; age; some drugs Fed state | Reduced absorption Slower rate of absorption Reduced solubility of basic drugs |
| Distribution | Increased body fat relative total body water Reduced albumin Increased AGP | Obesity Liver disease Obesity | Increased volume of distribution of lipophilic drug. Reduces protein binding of acidic drugs and increases that of basic drugs. Appropriate changes to both drug distribution and metabolism. |
| Metabolism | Reduced CYP activity | Polymorphism Disease or age | Reduced metabolism |
| Elimination | Reduced GFR and tubular functions | Age | Altered elimination of drugs that are predominantly cleared by the kidney. For compounds that are glucuronidated, the parent drug recirculates for longer due to reduced elimination of the glucuronide. |
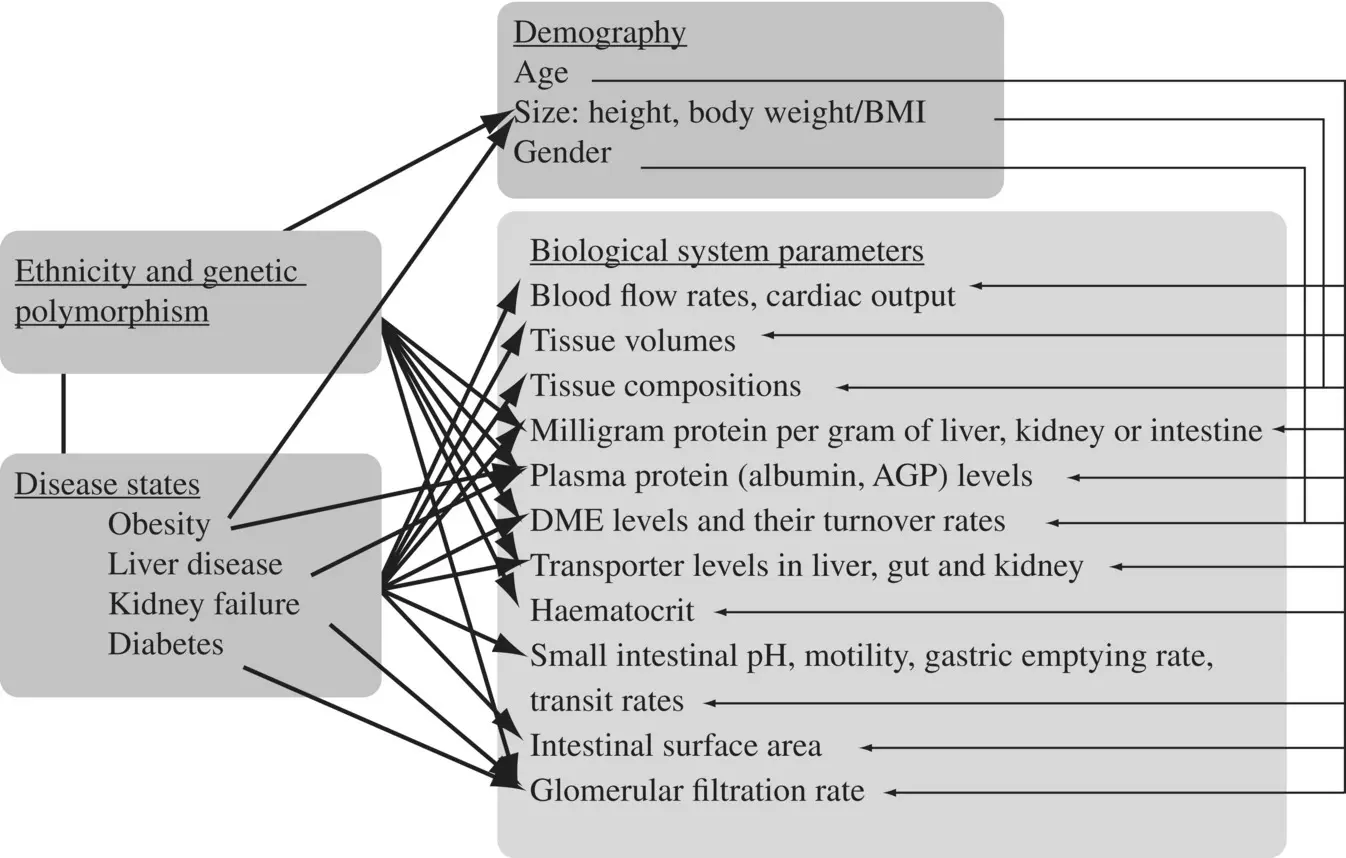
Figure 1.11. Sources of variability in the physiological parameters that impact pharmacokinetics.
1.4 PHARMACOKINETICS OPTIMIZATION IN DRUG DISCOVERY
A successful lead optimization of PK aims at maximizing the bioavailability and half‐life, reducing clearance and toxic metabolites and minimizing the risk for drug–drug and food–drug interactions. A comprehensive preclinical pharmacokinetic evaluation would ensure that compounds do not fail in the clinic (Singh, 2006). Table 1.4summarizes the PK optimizations and describes how and why they should be done.
1.5 PHARMACODYNAMIC PRINCIPLES
Pharmacokinetics provides an understanding of factors affecting absorption, distribution, metabolism, and excretion of an administered drug, all of which determine its exposure or concentration at the target organ (effect site). Relation of this exposure to the onset, intensity and duration of drug action is determined by pharmacodynamics. As Leslie Benet stated succinctly, “pharmacokinetics may be simply defined as what the body does to the drug, as opposed to pharmacodynamics which may be defined as what the drug does to the body”. A well‐defined, quantitative relationship between drug concentrations in biological fluids and pharmacodynamic effect provides the basis for defining a dosing regimen.
TABLE 1.4. Optimization of pharmacokinetics – what? how? and why? (Source: Singh, 2006)
| PK optimization | How? | Need for optimization |
|---|---|---|
| Gut bioavailability | Balance lipophilicity and solubility to achieve good absorption. Reduce potential for CYP3A metabolism and glucuronidation, the 2 major pathways for gut extraction. | To enhance bioavailability. |
| Clearance | Low clearance can be achieved by reducing lipophilicity, by avoiding functional groups that are known targets for metabolism and by reducing the activity of potential sites of metabolism through steric hindrance . Avoid reliance on single elimination pathway especially the high affinity, low capacity CYPs (2C9 and 2C19) and CYP3A4 (common pathway for many drugs) Avoid clearance through polymorphic enzymes (like CYP2D6) or transporters (OATP1B1). | Reduce hepatic extraction. Reduce potential for being a victim of DDI. Minimize the inter‐individual variability in exposure. |
| Volume of distribution | The greater the lipophilicity and greater the fraction unbound in plasma, the greater the V ss. Bases generally have a high V ss, followed by neutrals and then acids. | Ensures long duration of the drug in the body. |
| Half‐life | A large volume of distribution and low clearance will ensure a long half‐life. | Long post‐dose duration will ensure a simplified dosing regimen of once daily and promote patient compliance. |
| Biotransformation | Avoid carboxylic acids that are likely to form reactive acyl glucuronides. Avoid reactive metabolites. | To reduce toxic effects. |
| Transporters | Ensure sufficient permeability to reduce interplay of transporter and metabolism. | To reduce potential for DDI. |
| Pharmacokinetics | Aim for linear PK by keeping the dose as low as possible. | To minimize uncertainty in predictions of disposition. |
1.5.1 Pharmacological Targets and Drug Action
A pharmacological effect often involves the modulation of an intrinsic physiological process by a drug that binds to a target protein. Pharmacological targets can be receptors and proteins involved in regulatory pathways, enzymes, structural proteins, nuclear receptors, transporter proteins, or ion channels ( Table 1.5). Drugs may be classified depending on how they modulate their receptor and achieve drug effect. A drug is said to be an agonist ( Figure 1.12) or partial agonist ( Figure 1.13) if it binds to a receptor and functionally mimics or enhances the action of an endogenous ligand. by stimulating receptor activity fully or partially. Partial agonism may be explained by either an inefficient modulation of receptor conformation/transduction following receptor occupancy or by a higher drug affinity to the inactive form of a receptor, that isomerizes between an active and an inactive form. A drug that reduces receptor activity through competing with the endogenous agonist for the same active site is called a competitive antagonist . A drug is said to be a noncompetitive antagonist if it blocks or inhibits the pharmacological response of an endogenous agonist, by binding to a different site and triggering a conformational change in the receptor protein. A drug that binds to a target irreversibly (e.g., covalent binding) is called an irreversible inhibitor . Both noncompetitive and irreversible inhibitors decrease the receptor count that is available for the endogenous agonist, thereby reducing its efficacy. Inverse agonists appear to act in an opposite manner to agonists at a receptor site. Unlike an agonist which increases receptor activity, or an antagonist which blocks its activity, an inverse agonist reverses the activity of constitutively active receptors that are coupled to second messenger pathways even in the absence of an agonist. The binding of a drug to a target protein can initiate several direct or indirect responses depending on the type of target protein, its location, and target class ( Figure 1.14). Most enzyme inhibitions lead to direct effect. Examples of indirect drug responses include the release of hormones and/or other endogenous ligands to stimulate a particular response or the activation of a second messenger which, in turn, initiates a series of biochemical reactions (e.g., drug binding to a G‐protein coupled receptor, GPCR) leading to the desired response.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»
Представляем Вашему вниманию похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.