Handbook of Aggregation-Induced Emission, Volume 1
Здесь есть возможность читать онлайн «Handbook of Aggregation-Induced Emission, Volume 1» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Handbook of Aggregation-Induced Emission, Volume 1
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Handbook of Aggregation-Induced Emission, Volume 1: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Handbook of Aggregation-Induced Emission, Volume 1»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
A thorough introduction to the mechanistic understanding of the importance of molecular motion to aggregation-induced emission An exploration of the aggregation-induced emission mechanism at the molecular level Practical discussions of aggregation-induced emission from the restriction of double bond rotation at the excited state, and clusterization-triggered emission Perfect for academic researchers working on aggregation-induced emission, this set of volumes is also ideal for professionals and students in the fields of photophysics, photochemistry, materials science, optoelectronic materials, synthetic organic chemistry, macromolecular chemistry, polymer science, and biological sciences.
Handbook of Aggregation-Induced Emission, Volume 1 — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Handbook of Aggregation-Induced Emission, Volume 1», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
2.3.3 Bending Vibration of Bonds
(CAAC Ad)CuCl (see Figure 2.3) is a two‐coordinate Cu(I) complex, which exhibits highly efficient fluorescence in aggregate [50]. The excited‐state decay dynamics for (CAAC Ad)CuCl in both solution and solid states is studied by the hybrid QM/MM approach, coupled with the TVCF rate formalism [42]. Analyzing the geometrical changes between S 0and S 1upon excitation, it is found that the complex is more flexible in solution. The largest changes appear in the coordination bond angles between copper and two ligands with ∠C 1−Cu−Cl, ∠Cu−C 1−N, and ∠Cu−C 1−C 4decreasing from 9.67°, 12.82°, and 10.73° in solution to 3.66°, 4.46°, and 6.75° in the solid phase, respectively. And the transition character changes from metal‐to‐ligand charge transfer (MLCT) to hybrid MLCT and halogen‐to‐ligand charge transfer (XLCT) upon aggregation. The k rare close in both phases, while k icdecreases by about 3 orders of magnitude from 8.38 × 10 7to 1.47 × 10 4s −1(see Table 2.2). The λ totaldecreases a lot from solution (7121 cm −1) to solid state (2274 cm −1). It is found that the largest contributions to λ totalcome from several low‐frequency bending modes and one high‐frequency stretching mode in solution. These low‐frequency modes are assigned to be the bending vibrations associated with coordination bonds C 1−Cu and Cu−Cl, and the high‐frequency mode belongs to the stretching vibration of the C−N bond in carbene ligand. Upon aggregation, the vibrations of angles C 1−Cu−Cl and Cu–C1–N are restricted largely, while the stretching vibrations of the C 1−N bond are insensitive to the environment ( Figure 2.4c). Overall, the strong solid‐state fluorescence of (CAAC Ad)CuCl is induced by removing the nonradiative decay channels owing to the decoupling between transition electron and the bending vibrations of coordination bonds (see Figure 2.2d).
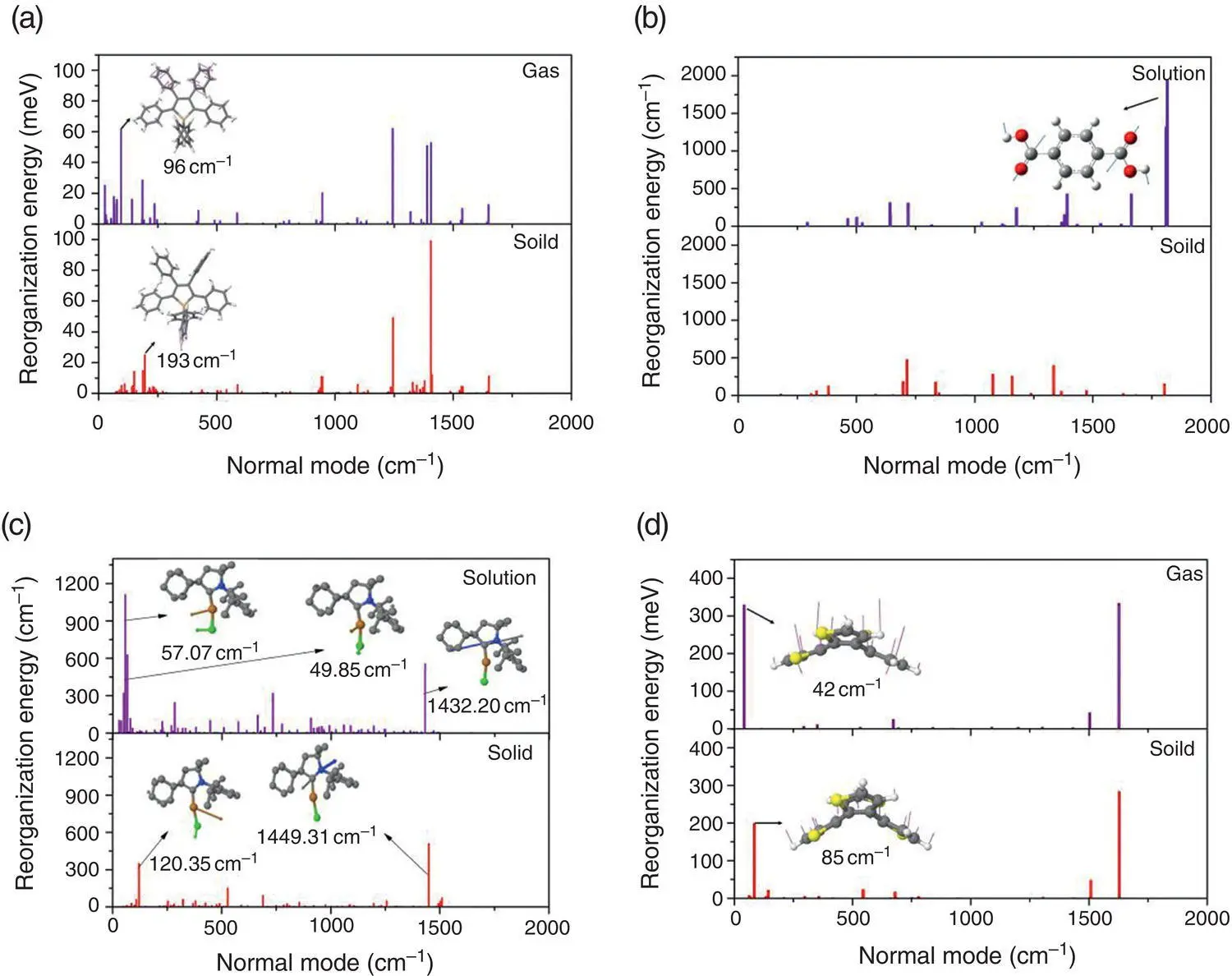
Figure 2.4 Calculated reorganization energies versus normal mode of (a) HPS, (b) TPA, (c) (CAAC Ad)CuCl, and (d) COTh in both gas/solution and solid phases, respectively.
2.3.4 Flipping Vibrations of Molecular Skeletons
Cyclooctotetraene (COT) is a prototype molecule with nonaromatic annulenes [51–53]. The COT derivative cyclooctatetrathiophene (COTh) was found to be AIE active and its AIE mechanism was further investigated theoretically and experimentally [43]. The theoretically optimized geometries in the S 0and S 1states show that the isolated COTh is much more flexible than that in a cluster. For example, from the S 0to S 1state, the modifications of the dihedral angles Θ I–IIand Θ II–III(I, II, and III denote the aromatic rings marked in Figure 2.3) are 22.82° and 24.22° in the gas phase, much larger than those in the solid state of 12.78° and 12.96°. Upon excitation, the dihedral angles between the neighboring thienyl rings are decreased from S 0to S 1. As shown in Table 2.3, for example, Θ I–IIdecreases from 44.93° to 22.11° in the gas phase, while it reduces from 43.84° to 31.06° in the solid phase, indicating a better planarity of the central large eight‐membrane ring. Therefore, the conjugation of the central eight‐membrane improves and the oscillator strength of S 1enhances accordingly. In addition, the excitation energy at the S 1geometry increases from 2.42 eV in the gas phase to 2.69 eV in cluster. Therefore, k rincreases from 9.80 × 10 4to 6.05 × 10 5s −1, owing to the enhancement of oscillator strength and excitation energy; see Table 2.2. The k icis closely related to the reorganization energy. For COTh, the λ totaldecreases to 636 meV in crystal from 860 meV in the gas phase and then the k icdecreases by 2 orders of magnitude. Thus, the Φ Fin the crystal is 3 orders of magnitude larger than that in the isolated state. As shown in Figure 2.4d, the major differences in the reorganization energies between the gas phase and the solid state are related to the low‐frequency eight‐member annulene flipping vibrational mode. Thus, the fluorescence of COTh upon aggregation is induced by the suppression of the nonradiative decay channel contributed by the electron–vibration coupling between the transition electron and low‐frequency flipping vibration (see Figure 2.2e).
Table 2.2 Calculated k r, k ic, and Φ Fin both solution and aggregate phases at room temperature for TPA, (CAAC Ad)CuCl, and COTh, respectively.
| k r(s −1) | k ic(s −1) | Φ F(%) | k r(s −1) | k ic(s −1) | Φ F(%) | |
|---|---|---|---|---|---|---|
| In isolated state | In solid phase | |||||
| TPA [41] | 3.34 × 10 4 | 4.97 × 10 7 | 0.067 | 3.43 × 10 7 | 5.15 × 10 6 | 86.95 |
| (CAAC Ad)CuCl [42] | 6.26 × 10 5 | 8.38 × 10 7 | 0.44 | 7.83 × 10 5 | 1.47 × 10 4 | 98.0 |
| COTh [43] | 9.80 × 10 4 | 4.13 × 10 9 | 0.002 | 6.05 × 10 5 | 1.87 × 10 7 | 3.13 |
2.3.5 Twisting Vibration of Molecular Skeletons
Coumarin (chromen‐2‐one) is a natural organic substance that is abundant in many plants. CD‐7 ( Figure 2.3) is a coumarin derivative with a seven‐membered aliphatic ring; it has typical AIE characteristics, while its analogue with a five‐membered aliphatic ring (CD‐5; Figure 2.3) exhibited an opposite ACQ effect [44]. The electronic structures in S 0and S 1at the level of (TD) M062X/6‐31G* were performed. The structural changes between S 0and S 1of CD‐7 are obvious. During the transition process from S 0to S 1, the mean torsion angle between the pyridinone and chromen‐2‐one fragments changes about 18°, indicating that CD‐7 undergoes out‐of‐plane twisting motions along the C 1–C 2bond under photoexcitation, while the corresponding geometric changes in the solid phase are only 5.8°, which suggests that the intramolecular twisting motion is largely restricted. In sharp contrast, CD‐5 has a more planar conformation of the conjugated framework in both the S 0and S 1and changes little in the gas phase, indicating its intrinsic structural rigidity. In addition, λ totalof CD‐7 reduced from 4028 cm −1in solution to 3438 cm −1in aggregates, owing to the restriction of twisting motion in aggregates. Thus, the restriction of twisting motion in aggregates blocks the nonradiative decay channels and enables CD‐7 to fluoresce strongly (see Figure 2.2f) [44].
Table 2.3 Calculation structural parameters of COTh [43] in both gas phase and the crystal (in degree).
| S 0 | S 1 | ∣∆(S 0− S 1)∣ | S 0 | S 1 | ∣∆(S 0− S 1)∣ | Crystal | |
|---|---|---|---|---|---|---|---|
| In gas phase | In solid phase | ||||||
| Θ I–II | 44.93 | 22.11 | 22.82 | 43.84 | 31.06 | 12.78 | 42.85 |
| Θ II–III | −48.58 | −24.36 | 24.22 | −48.67 | −35.71 | 12.96 | −48.83 |
| Θ III–IV | 44.92 | 22.11 | 22.81 | 45.24 | 32.60 | 12.64 | 45.16 |
| Θ I–IV | −48.59 | −24.36 | 24.23 | −48.94 | −35.72 | 13.22 | −46.99 |
THBDBA contains two pairs of phenyl rings that are covalently linked through a singly bonded ethyl chain ( Figure 2.3); it is almost nonemissive when fully solvated in the THF solution and starts to emit with the addition of water [9]. In THBDBA crystal, each set of phenyl rings locked by the ethyl chain adopts a V‐shaped or “boat” conformation. Theoretical calculation indicates that the single molecule has a λ totalof 6898.6 cm −1, whereas the molecule in a cluster only has 5901.5 cm −1. The normal mode analysis shows that low‐frequency vibrational modes mainly contribute to λ totalfor both single molecule and in crystal, but λ totalin the crystal is largely reduced. Upon aggregation, the restriction of low‐frequency twisting motion blocks the nonradiative decay channel and turns the fluorescence on ( Figure 2.2g) [9].
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Handbook of Aggregation-Induced Emission, Volume 1»
Представляем Вашему вниманию похожие книги на «Handbook of Aggregation-Induced Emission, Volume 1» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Handbook of Aggregation-Induced Emission, Volume 1» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.