Handbook of Aggregation-Induced Emission, Volume 1
Здесь есть возможность читать онлайн «Handbook of Aggregation-Induced Emission, Volume 1» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Handbook of Aggregation-Induced Emission, Volume 1
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Handbook of Aggregation-Induced Emission, Volume 1: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Handbook of Aggregation-Induced Emission, Volume 1»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
A thorough introduction to the mechanistic understanding of the importance of molecular motion to aggregation-induced emission An exploration of the aggregation-induced emission mechanism at the molecular level Practical discussions of aggregation-induced emission from the restriction of double bond rotation at the excited state, and clusterization-triggered emission Perfect for academic researchers working on aggregation-induced emission, this set of volumes is also ideal for professionals and students in the fields of photophysics, photochemistry, materials science, optoelectronic materials, synthetic organic chemistry, macromolecular chemistry, polymer science, and biological sciences.
Handbook of Aggregation-Induced Emission, Volume 1 — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Handbook of Aggregation-Induced Emission, Volume 1», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
2.3.1 Rotating Vibrations of Intramolecular Aromatic Ring
Siloles (silacyclopentadines) are the first kind of AIEgens [4, 45, 46]. The first silole molecule where the Φ Fin both gas phase and crystal are quantitatively investigated is HPS ( Figure 2.3) [37]. HPS is poorly luminescent in cyclohexane at room temperature with a Φ Fas low as 0.30%, while in thin film, the Φ Freaches 78% [45]. Combining QM and QM/MM calculations, the structural change analysis of optimized structures between S 0and S 1reflects that the torsional angles between the central silacycle and phenyl groups at the 2,5‐positions show larger modifications (14.56° and 14.63°) in the isolated state than those (5.85° and 1.05°) in solid phase, suggesting that the geometric relaxations of the phenyl rings at the 2,5‐positions are largely hindered in the solid phase. Moreover, the dihedral angle between the central silacycle and the phenyl ring at the 5‐position decreases, which strengthens the intramolecular conjugation, resulting in the increase of the electric transition dipole moment ( μ ) from 5.20 to 5.85 Debye ( Table 2.1). Hence, upon aggregation, the calculated k rincreases 6 times, while k icdecreases about 4 orders of magnitude ( Table 2.1). The nonradiative decay channels are generated by the coupling between the electrons and thermal vibrations that can be quantified by the molecular reorganization energy 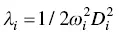 for the i th vibrational mode and
for the i th vibrational mode and 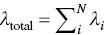 , which is very sensitive to the surrounding environment. As seen, λ totalfor HPS in the solid phase (403 meV) is obviously smaller than that in the gas phase (492 meV). The decrease in λ totalmainly stems from the low‐frequency vibrational modes (see Figure 2.4a), which are assigned to the rotating vibrations of the phenyl rings at the 2,5‐position. Thus, the excited‐state energy dissipation pathways are easily restricted via the decoupling between the electron and low‐frequency out‐of‐plane rotating modes in aggregates (see Figure 2.2b). The corresponding theoretically calculated Φ Fin the gas phase and solid phases is 0.003 and 76% (see Table 2.1), respectively, consistent with experimental results [45]. This well explains the bright emission of HPS in aggregate phase.
, which is very sensitive to the surrounding environment. As seen, λ totalfor HPS in the solid phase (403 meV) is obviously smaller than that in the gas phase (492 meV). The decrease in λ totalmainly stems from the low‐frequency vibrational modes (see Figure 2.4a), which are assigned to the rotating vibrations of the phenyl rings at the 2,5‐position. Thus, the excited‐state energy dissipation pathways are easily restricted via the decoupling between the electron and low‐frequency out‐of‐plane rotating modes in aggregates (see Figure 2.2b). The corresponding theoretically calculated Φ Fin the gas phase and solid phases is 0.003 and 76% (see Table 2.1), respectively, consistent with experimental results [45]. This well explains the bright emission of HPS in aggregate phase.
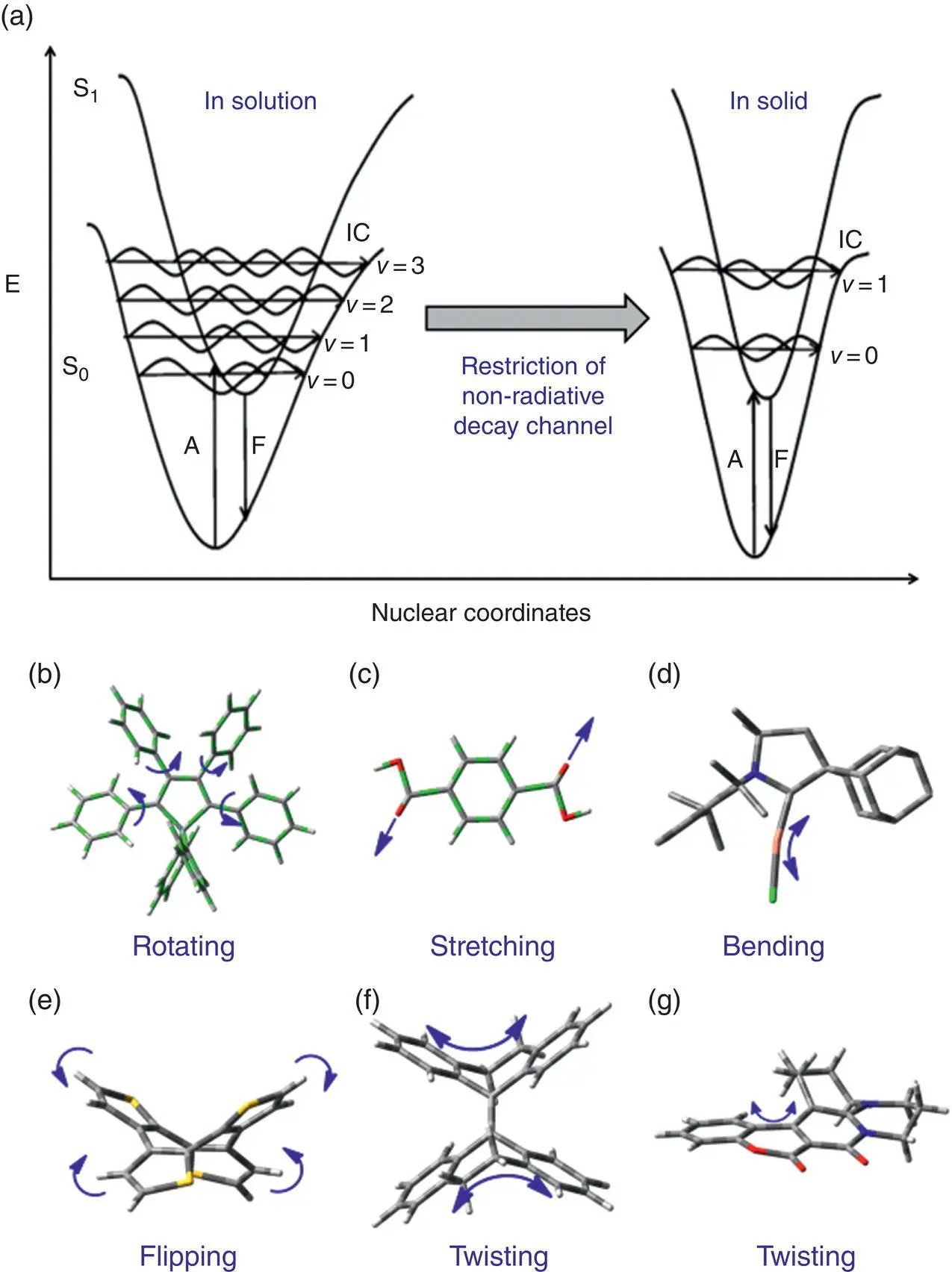
Figure 2.2 (a) PES scheme of AIEgens in solution and solid states. (b–g) Overview of vibrational modes involved in the excited‐state nonradiative decay channels, which contribute to AIE.
A similar AIE mechanism is followed by the other siloles, with the degree of π ‐conjugation of the 2,5‐substituents increasing from TPS [47], BrTPS [48], HPS [45], BTPES [48] to BFTPS [49] ( Figure 2.3). As shown in Table 2.1, as the conjugation increases, ∆ E gin both gas phase and solid state decreases regularly. Simultaneously, the μ value increases drastically due to the electron delocalization. The balance of excitation energy and μ makes k rto first increase and then levels off as the extension of conjugation from TPS to BFTPS. After aggregation, λ totalfor all AIEgens decreases obviously and k icdecreases by more than 2 orders of magnitude from 9.30 × 10 5s −1for TPS to 1.22 × 10 8s −1for BFTPS. Overall, with the degree of π ‐conjugation of the 2,5‐substituent increases, Φ Fin the solid state first increases sharply, then levels off, and finally starts to decrease slightly, as the competition between k rand k ic. Similar to the above discussed HPS, the dramatic decrease of k icupon aggregation turns the fluorescence on, which is mainly caused by the decoupling of the electron and low‐frequency rotational normal modes ( Figure 2.4a) [38].
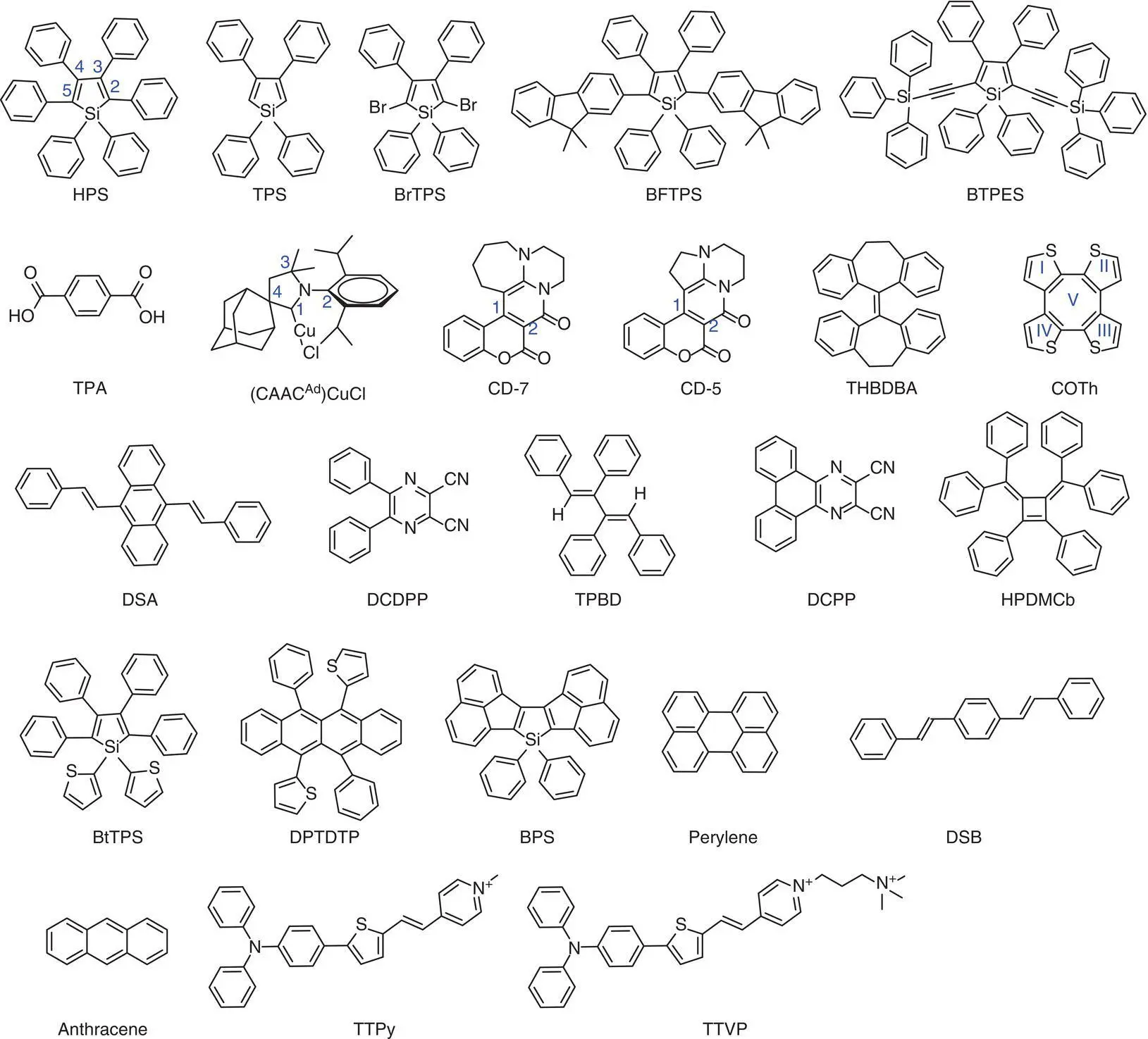
Figure 2.3 Overview of molecular structures discussed in Sections 2.3and 2.4.
2.3.2 Stretching Vibrations of Bonds
Terephthalic acid (TPA) is a representative AIEgen, which could emit both fluorescence and phosphorescence simultaneously [41]. Through a combined QM/MM approach, the photophysical properties of TPA in both gas phase and crystal were investigated to unravel the effect of crystallization on the nature of the molecular excited states. It is found that the nature of S 1changes from ( n , π *) in the gas phase to ( π , π *) in crystal because the excitation energy of ( n , π *) is increased up to 5.05 eV from 4.81 eV, whereas the ( π , π *) state is reduced to 4.76 eV from 4.99 eV due to the strong electrostatic interaction upon aggregation. Accordingly, the oscillator strength of S 1is largely enhanced to 3.49 × 10 −2in the crystalline phase from 3.33 × 10 −5in the gas phase, which recovers the radiative decay from S 1to S 0. And the resultant k ris largely increased by 3 orders of magnitude from 3.34 × 10 4s −1in the gas phase to 3.43×10 7s −1in the solid phase.
Table 2.1 Calculated HOMO–LUMO energy gap (∆ E g), electric transition dipole moment ( μ ), total reorganization energy ( λ total), k r, k ic, and Φ Fin gas phase and aggregate phase at room temperature for HPS, TPS, BrTPS, BTPES, and BFTPS, respectively.
| ∆ E g(eV) | μ (Debye) | λ total(meV) | k r(s −1) | k ic(s −1) | Φ F(%) | |
|---|---|---|---|---|---|---|
| In gas phase | ||||||
| HPS [37] | 3.59 | 5.20 | 492 | 1.05 × 10 7 | 3.76 × 10 11 | 0.003 |
| TPS [38] | 3.21 | 0.58 | 1120 | 9.30 × 10 5 | 1.62 × 10 10 | 0.01 |
| BrTPS [38] | 2.90 | 1.71 | 1161 | 5.55 × 10 6 | 5.72 × 10 9 | 0.09 |
| HPS [38] | 2.62 | 5.26 | 891 | 4.98 × 10 7 | 2.53 × 10 10 | 0.20 |
| BTPES [38] | 2.48 | 6.03 | 667 | 6.76 × 10 7 | 2.66 × 10 10 | 0.25 |
| BFTPS [38] | 2.36 | 9.54 | 821 | 1.22 × 10 8 | 1.66 × 10 9 | 6.86 |
| In solid phase | ||||||
| HPS [37] | 3.48 | 5.85 | 403 | 6.56 × 10 7 | 2.06 × 10 7 | 76.0 |
| TPS [38] | 3.26 | 0.60 | 932 | 1.15 × 10 6 | 3.32 × 10 6 | 25.8 |
| BrTPS [38] | 2.91 | 1.80 | 890 | 8.12 × 10 6 | 1.50 × 10 6 | 82.3 |
| HPS [38] | 2.70 | 5.90 | 753 | 7.43 × 10 7 | 1.57 × 10 6 | 97.9 |
| BTPES [38] | 2.49 | 5.75 | 592 | 6.57 × 10 7 | 1.93 × 10 6 | 97.1 |
| BFTPS [38] | 2.30 | 9.10 | 607 | 1.14 × 10 8 | 1.07 × 10 7 | 91.4 |
The reorganization energy analysis from S 1→ S 0process for each normal mode in the gas phase reflects that the C=O stretching vibration contributes the most (∼1974.37 cm −1), consistent with the corresponding large structural modification (0.08 Å between S 0and S 1). However, in the solid phase, this value is reduced to 212.96 cm −1and the corresponding structural change is only 0.01 Å. Such a remarkable reduction is due to the alternation to π → π * electronic transition, which is decoupled with the C=O stretching vibration. As a result, from the gas to solid phases, k icdecreases about 1 order of magnitude from 4.97 × 10 7to 5.15 × 10 6s −1. That is, the nonradiative decay process is blocked by the decoupling between the high‐frequency C=O stretching vibration and transition electrons ( Figures 2.2c and 2.4b). Overall, the largely accelerated radiative decay rate and slowed nonradiative decay rate induce the observed strong fluorescence in the solid phase [41].
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Handbook of Aggregation-Induced Emission, Volume 1»
Представляем Вашему вниманию похожие книги на «Handbook of Aggregation-Induced Emission, Volume 1» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Handbook of Aggregation-Induced Emission, Volume 1» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.