Brenda A. Wilson - Bacterial Pathogenesis
Здесь есть возможность читать онлайн «Brenda A. Wilson - Bacterial Pathogenesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Bacterial Pathogenesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Bacterial Pathogenesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Bacterial Pathogenesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Completely revised and updated, and for the first time in stunning full-color, Bacterial Pathogenesis: A Molecular Approach, Fourth Edition, builds on the core principles and foundations of its predecessors while expanding into new concepts, key findings, and cutting-edge research, including new developments in the areas of the microbiome and CRISPR as well as the growing challenges of antimicrobial resistance. All-new detailed illustrations help students clearly understand important concepts and mechanisms of the complex interplay between bacterial pathogens and their hosts. Study questions at the end of each chapter challenge students to delve more deeply into the topics covered, and hone their skills in reading, interpreting, and analyzing data, as well as devising their own experiments. A detailed glossary defines and expands on key terms highlighted throughout the book. Written for advanced undergraduate, graduate, and professional students in microbiology, bacteriology, and pathogenesis, this text is a must-have for anyone looking for a greater understanding of virulence mechanisms across the breadth of bacterial pathogens.
Bacterial Pathogenesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Bacterial Pathogenesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
For example, the Illumina sequencing technology is routinely used to locate mutations in bacteria, such as Streptococcus pneumoniae, whose genome contains about 2.2 million base pairs. In this case, chromosomal DNA isolated from mutant strains is sheared into random fragments of about 500 base pairs. Adaptors required for hybridization during the sequencing method are ligated to the ends of the DNA fragments, and the resulting products are amplified by PCR to give random libraries of genomic fragments from each mutant. The adaptors used for each mutant genome have slightly different sequences (“barcodes”), so that DNA sequences from more than one mutant can be sequenced simultaneously and later distinguished. The resulting barcoded libraries are mixed and subjected to Illumina sequencing. Currently, with the highest-capacity instruments, such a sequencing run yields about 120 billion bases of sequence comprised of approximately 400 million reads with lengths of up to 300 bases! Thus, the base-pair coverage of a single S. pneumoniae genome in a reaction lane would be about 27,000-fold in this example. This extraordinarily high level of coverage means that barcoded, fragmented genomic libraries from 27 different mutants could be sequenced simultaneously at 1,000-fold coverage in a single sequencing run in less than a week. Subsequent bioinformatic sequence comparisons with the known genome sequence of S. pneumoniae could then reveal the locations of single-nucleotide point mutations, small and large deletions, or even regions of chromosomal duplication that contribute to the mutant phenotypes. And what is the cost? At this writing, the cost of the sequence determination and bioinformatic analyses per mutant genome is far less than the cost of classical genetic approaches, which previously required weeks to months to obtain and, if they worked, would yield far less information compared to less than one week for the large-scale sequencing analysis.
16S rRNA Gene-Based Profiling of Complex Microbial Communities. Scientists are often interested in understanding the effect of certain conditions or factors over time on composition of the microbial community. For instance, a researcher might be interested in the effect of diet, hormones, or age on the composition of the microbiota, or how antibiotic therapy impacts the microbiota. To accomplish this, it is necessary to collect at any given time a sample of the microbes that is representative of the whole community (a profile) such that changes in abundance or diversity can be monitored.
One profiling method involves DNA microarray chips, called phylochips, which are comprised of tens of thousands of oligonucleotide-containing spots, each corresponding to a set of taxa-defining sequences, including rRNA gene regions and other unique gene sequences that can distinguish among the various microbial species (taxa or “phylotypes”) present in the sample of interest ( Figure 5-8). To design appropriate phylochips, the microbial species present in environmental niches, including different areas of the human body, must first be identified by rRNA gene sequencing methods from a representative sample. Oligonucleotide probes that will hybridize to the rRNA genes from each species are then printed onto the microarray. These phylochips can be used to monitor shifts in microbial community compositions in environmental and clinical samples. The greatest advantage of this phylochip technology is that all microorganisms of interest in an entire community, not just bacteria, can be detected in a single assay by multiple probes to give reliable taxonomic information.
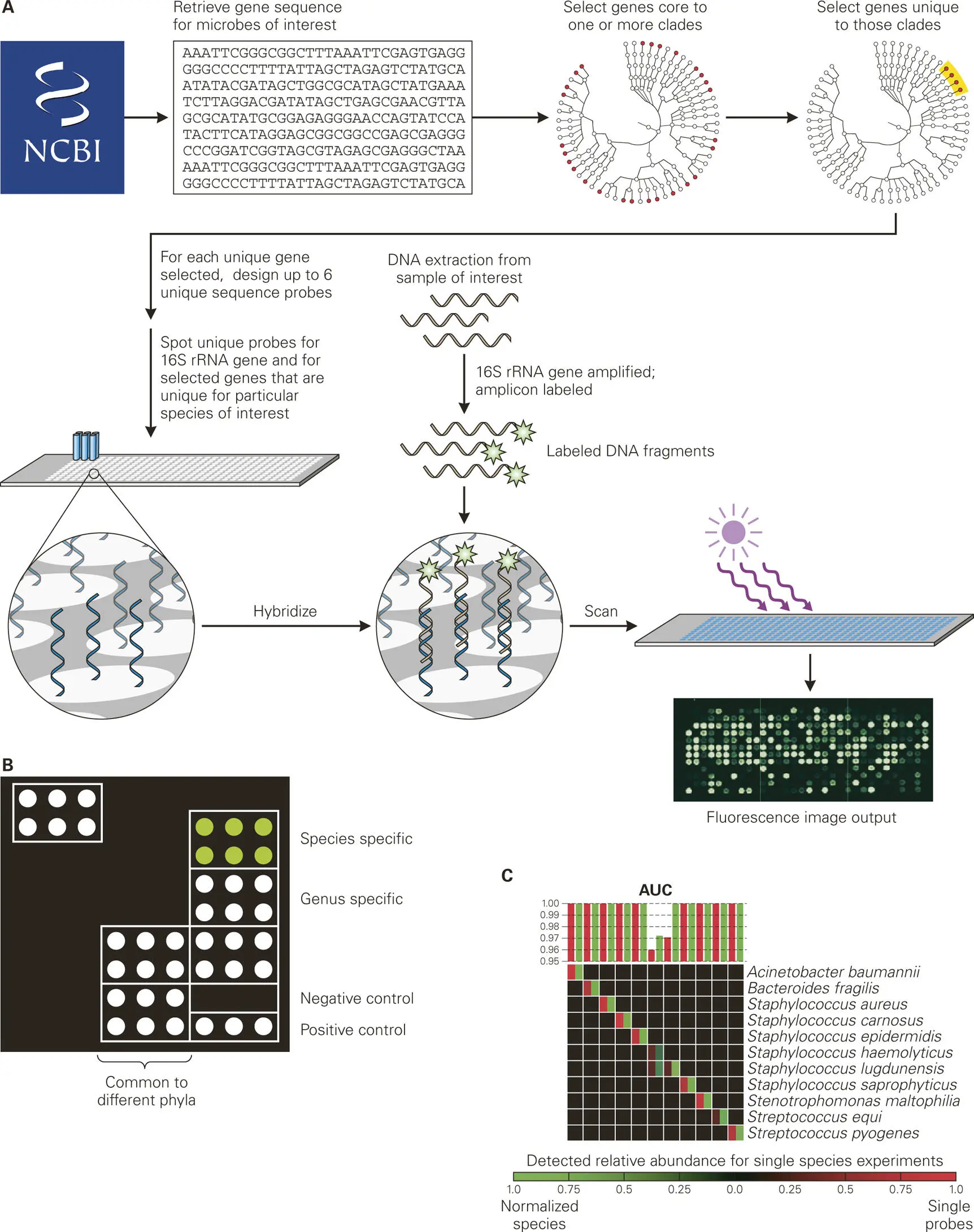
Figure 5-8. Phylochips for microbial-community profiling. Current phylochip platforms are a microarray-based method that taxonomically identifies microbes in a given sample through hybridization of the rRNA gene in that sample to all nine of the variable regions of the 16S rRNA gene. (A) General procedure for making a phylochip. Complete genomes are retrieved from the National Center for Biotechnology Information microbial database and a phylogenetic relationship tree is generated. Genome comparison of bacteria within each clade (group of related bacteria) is used to identify core gene sequences that are shared by most members of the clade. After sequence alignment with all other bacterial and archaeal genomes, core gene sequences that are unique to each target clade (species) are identified, and DNA probes are designed against up to 10 of the unique genes for each target species. (B) Example of how a phylochip could be used to distinguish microbes at the genus and species levels in a sample. Positive and negative controls are included to ensure that the hybridization steps worked and that there is no background detection, respectively. Some spots are probes targeted toward distinguishing specific microbes at the genus level (nine spots with probes for each of the V1–V9 variable regions of the 16S rRNA genes), while other spots are probes targeted toward distinguishing specific microbes at the species or subspecies level (spots corresponding to unique genes found only in particular clades or subspecies). Custom probes (upper left corner of figure) can be made for identifying species with particular genes present (e.g., virulence factors). (C) Heat map showing the intensities for individual probes (red) and total species probes (green) in the bacterial sample. A plot of the area under the curve (AUC) shows the relative abundances of each of the bacterial species in the sample.
The current limitation of these phylochips is that they first require the availability of the rRNA gene and other key distinguishing gene sequences for identification of each of the microbes expected to be present in the samples. But the increasing number of powerful high-throughput sequencing technologies currently available at ever more affordable costs, as well as the vast number of microbial sequences already deposited into the DNA databases, makes the design of phylochips for various microbial and clinical ecosystems increasingly feasible. Indeed, the breathtaking advances in massively parallel DNA sequencing methods described previously now allow cost-effective studies of the changing dynamics of complex microbial communities over time and under different conditions by direct sequencing alone.
Comparative 16S rRNA Gene-Based Profiling of Complex Microbial Communities. Current massively parallel sequencing technologies allow researchers to sequence millions of 16S rRNA genes per sample and multiple samples simultaneously, enabling microbial profiles for multiple samples to be rapidly generated and compared. Because of the depth of sequencing obtained from these large numbers of reads, these profiling methods enable researchers to rapidly assess differences in patterns between complex microbial communities, thereby gaining insight into the diversity or dynamics of the microbial communities.
It should be noted, however, that only nearly complete 16S rRNA sequences provide accurate measures of taxonomic identity and diversity within a microbial community. Unfortunately, while current sequencing technologies have made microbial community profiling routine and affordable, this has come at the expense of read length: sequence reads typically cover only a portion (usually 250–600 nucleotides) of the ∼1.5-kB 16S rRNA gene, necessitating that researchers choose shorter regions, such as V1–V3, V3–V4, V4–V5, or V3–V5 (see Figure 5-1A), for sequence analysis. Consequently, this compromises the resolution of the taxonomic identification and phylogenetic relationship determinations and should be taken into account when interpreting microbial sequencing data.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Bacterial Pathogenesis»
Представляем Вашему вниманию похожие книги на «Bacterial Pathogenesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Bacterial Pathogenesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.