Brenda A. Wilson - Bacterial Pathogenesis
Здесь есть возможность читать онлайн «Brenda A. Wilson - Bacterial Pathogenesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Bacterial Pathogenesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Bacterial Pathogenesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Bacterial Pathogenesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Completely revised and updated, and for the first time in stunning full-color, Bacterial Pathogenesis: A Molecular Approach, Fourth Edition, builds on the core principles and foundations of its predecessors while expanding into new concepts, key findings, and cutting-edge research, including new developments in the areas of the microbiome and CRISPR as well as the growing challenges of antimicrobial resistance. All-new detailed illustrations help students clearly understand important concepts and mechanisms of the complex interplay between bacterial pathogens and their hosts. Study questions at the end of each chapter challenge students to delve more deeply into the topics covered, and hone their skills in reading, interpreting, and analyzing data, as well as devising their own experiments. A detailed glossary defines and expands on key terms highlighted throughout the book. Written for advanced undergraduate, graduate, and professional students in microbiology, bacteriology, and pathogenesis, this text is a must-have for anyone looking for a greater understanding of virulence mechanisms across the breadth of bacterial pathogens.
Bacterial Pathogenesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Bacterial Pathogenesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
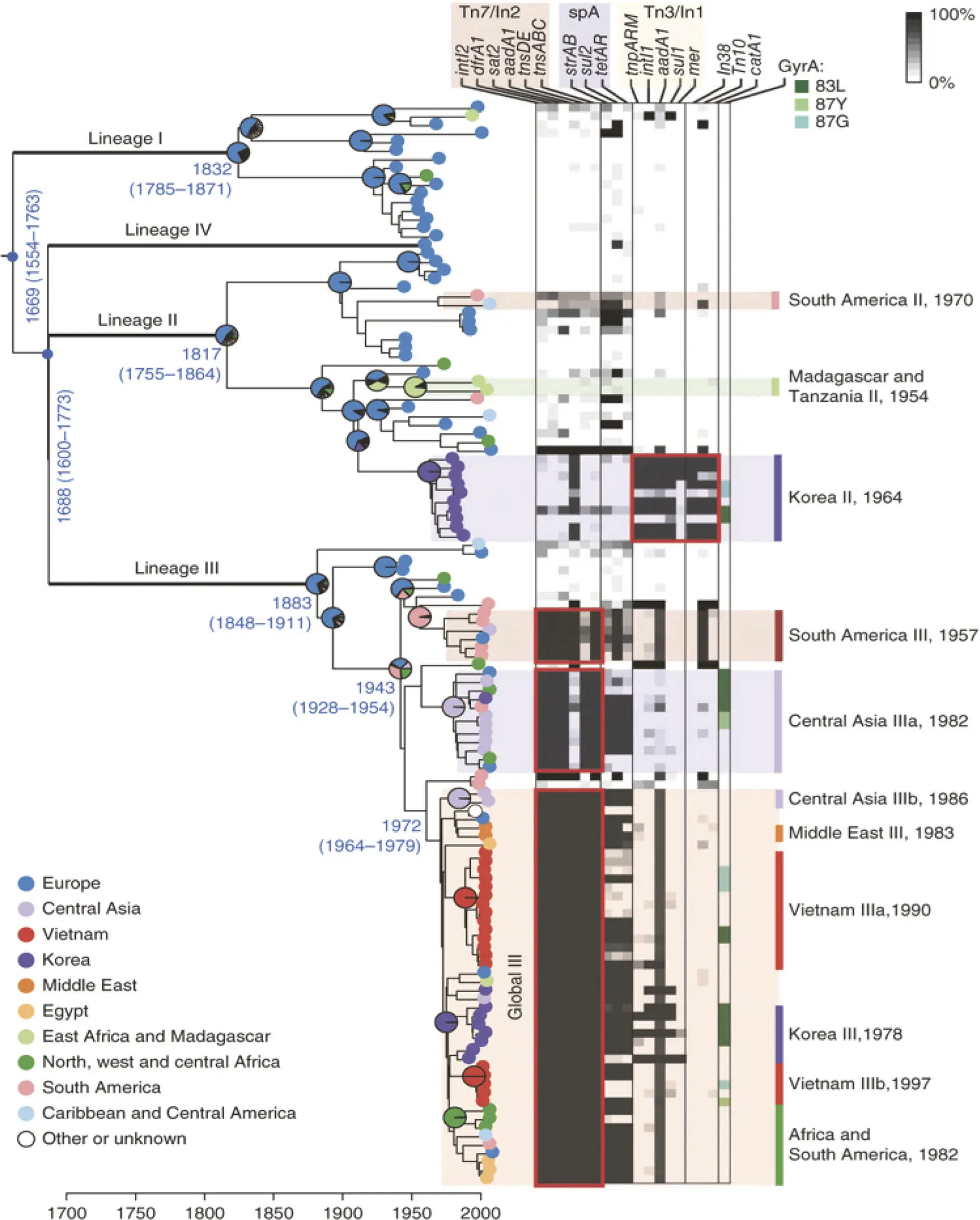
Figure 5-10. Phylogenetic relationship analysis using comparative whole-genome sequence profiling to track the evolution and dissemination of the dysentery pathogen Shigella sonnei. The dysentery pathogen Shigella sonnei was once predominant in developed countries, but it is now emerging as a major problem in developing countries. Whole-genome sequencing of 132 globally distributed clinical isolates, followed by phylogenetic analysis, showed that the current S. sonnei strains descended from a common ancestor in Europe less than 500 years ago. The results also showed that by the late 19th century, S. sonnei had diverged into four distinct lineages with strong regional clustering. The heat map shows the distribution of genes associated with antibiotic resistance. Known antibiotic-resistance mutations in the gene encoding DNA gyrase, GyrA, are indicated by color. Probable multidrug-resistance (MDR) gene acquisition events are boxed in red. Geographically localized clonal expansions are highlighted with their median estimated divergences dates. Reprinted from Holt KE, Baker S, Weill FX, Holmes EC, Kitchen A, Yu J, Sangal V, Brown DJ, Coia JE, Kim DW, Choi SY, Kim SH, da Silveira WD, Pickard DJ, Farrar JJ, Parkhill J, Dougan G, Thomson NR. 2012. Nat Genet 44:1056–1059, with permission.
Characterizing Microbiomes by Using Metagenomic Analysis
The various rRNA gene sequencing approaches mentioned previously only give information regarding what types of microbes are present in a community. A limitation of this type of approach is its failure to generate functional genomic information for deciphering the metabolic contributions of the microbes present in an ecosystem. For example, it does not provide direct information about which microbes might have a symbiotic relationship by producing and secreting metabolic products that could cross-feed other microbial species or the host. The current state of mega-scale DNA sequencing technology has spurred interest in a more ambitious approach to analyzing the body’s microbiota: determining the sequences of all of the individual microbial genomes of the body (the microbiomes). The goal of this approach, called metagenomic analysis, is designed to go beyond the question of cataloging what organisms are present (i.e., the census question) to the question of what the metabolic and physiologic potential of the microbiota is (i.e., the metabolic genes and pathways that are present).
But what can be done about species whose genomes are incomplete or not in the database at all? To answer this question, the next stage of metagenomic analysis involves isolation and mega-scale sequencing of all genomic DNA from an entire mixed microbial population (called the metagenome) so as to harvest the remarkable and vast diversity present. In fact, the advances in robotics, ultra-high-throughput, massively parallel sequencing, and bioinformatics assembly technologies are already leading to determination of complete genomes of microbes directly (without cultivation or cloning of individual isolates) from the mixed genomic DNA samples isolated from complex microbiota communities. For instance, metagenomic analysis can be applied to profiling strain-level variation in microbial communities ( Figure 5-11).
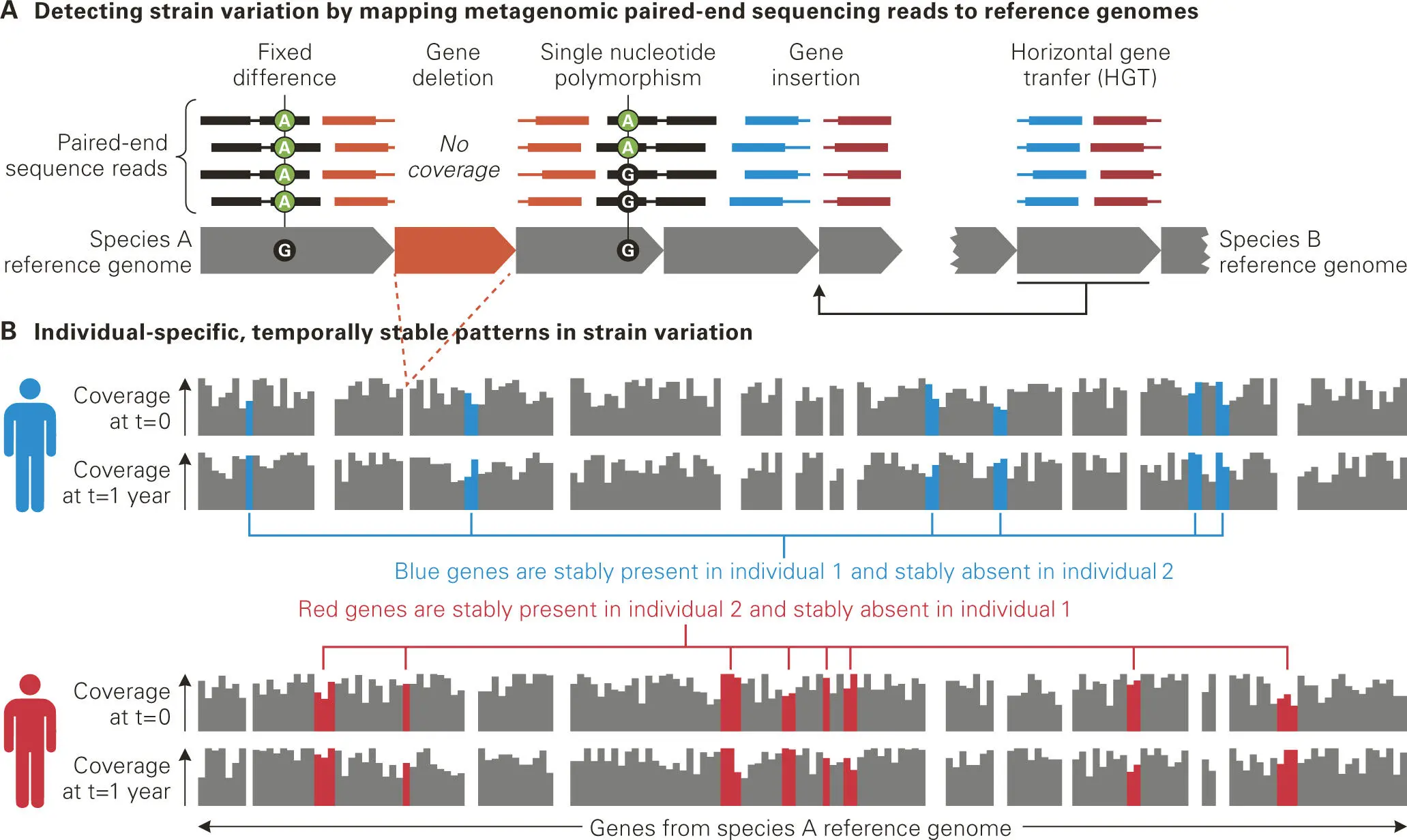
Figure 5-11. Metagenomic profiling of strain-level variation in microbial communities. (A) Mapping paired-end sequencing reads to microbial reference genomes reveals not only the genomes that are present in a community, but also differences between the isolates of particular species and the reference isolate. In this example, most positions have 4x coverage, represented by four sequencing reads mapped to each position in the reference genome sequences from bacterial species A and B. Gene deletion events can be detected with relatively low coverage of the reference genome; no reads from the sample map to deleted genes (in orange). Higher sequencing coverage of the genomes facilitates differentiating between sequencing error and true nucleotide-level strain variation. Such variation includes fixed differences (in which the sample is consistently different from the reference at some site) and single nucleotide polymorphisms (SNPs; in which a site occurs in two or more states in the sample). Sequence reads that do not map together (blue reads from individual 1 and red reads from individual 2) indicate additional community variation, including the insertion of genomic material not found in the reference genome by mechanisms such as horizontal gene transfer (HGT). (B) Mapping reads to reference genomes can reveal patterns of gene presence or absence, which is a form of strain variation. Here, two individuals sampled at two time points (t = 0 and t = 1 year) are distinguished by the presence or absence of genes in species A. The blue genes are stably present in individual 1 and stably absent in individual 2, whereas the red genes are stably present in individual 2 and stably absent in individual 1. Adapted from Franzosa EA, Hsu T, Sirota-Madi A, Shafquat A, Abu-Ali G, Morgan XC, Huttenhower C. 2015. Nat Rev Microbiol 13(6):360–372, with permission.
Assembling individual genome sequences from many thousands of sequences is still challenging for existing bioinformatics programs, but here again, rapid advances are being made in the analysis of the huge volume of new sequence data that is emerging, and these advances are beginning to make what seems unimaginable today feasible tomorrow (in biotechnology, as in most modern scientific endeavors, “impossible” just means it is “not possible yet”). Interpretation of a metagenomic analysis is much more complex than 16S rRNA analysis, or even a whole genome assembly and analysis, and details are beyond the scope of this textbook, but for the adventurous, several examples of recent metagenomic analyses of human microbiotas are provided in the suggested readings. What these analyses allow us to do is have a glance at the types of biosynthetic and metabolic pathways that the microbes in any given population might have at their disposal to utilize.
To help tackle the daunting task of defining our microbiomes, the National Institutes of Health launched the Human Microbiome Project (HMP) in 2007 with the objective of sequencing the collective genomes of all of the microbes (bacteria, archaea, fungi, protozoa, and viruses) that comprise the microbiotas of five targeted sites in the human body: the skin, mouth, nasal passages/lungs, vagina, and gut. The goal of the HMP was to lay the groundwork for establishing what constitutes a healthy microbiota so as to understand how changes in microbial composition, in terms of diversity and richness of content, affect health and disease. A large part of the HMP was to establish a repository of reference microbial genome sequences that could be used for facilitating the interrogation of microbiomes.
Metagenomic analysis starts with the census information indicating which species are present in microbial populations, but then adds information about metabolic function gleaned from thousands of complete bacterial genomes that are already deposited in genome databases, largely established through the HMP effort (see Box 5-1). These resources can provide a vast amount of information about the metabolic potential of a particular microbiome through comparison of the metabolic pathway sequences present in that mixed bacterial population with those from annotated genomes of taxonomically related bacteria that have been collected through the HMP. Since most of the already completed genomes were sequenced from a single clone of that microbe, most of the existing metabolic pathways and gene functions have already been ascertained, either directly from biochemical analyses or by inference based on analogy to similar genes in other microbes.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Bacterial Pathogenesis»
Представляем Вашему вниманию похожие книги на «Bacterial Pathogenesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Bacterial Pathogenesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.