Brenda A. Wilson - Bacterial Pathogenesis
Здесь есть возможность читать онлайн «Brenda A. Wilson - Bacterial Pathogenesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Bacterial Pathogenesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Bacterial Pathogenesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Bacterial Pathogenesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Completely revised and updated, and for the first time in stunning full-color, Bacterial Pathogenesis: A Molecular Approach, Fourth Edition, builds on the core principles and foundations of its predecessors while expanding into new concepts, key findings, and cutting-edge research, including new developments in the areas of the microbiome and CRISPR as well as the growing challenges of antimicrobial resistance. All-new detailed illustrations help students clearly understand important concepts and mechanisms of the complex interplay between bacterial pathogens and their hosts. Study questions at the end of each chapter challenge students to delve more deeply into the topics covered, and hone their skills in reading, interpreting, and analyzing data, as well as devising their own experiments. A detailed glossary defines and expands on key terms highlighted throughout the book. Written for advanced undergraduate, graduate, and professional students in microbiology, bacteriology, and pathogenesis, this text is a must-have for anyone looking for a greater understanding of virulence mechanisms across the breadth of bacterial pathogens.
Bacterial Pathogenesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Bacterial Pathogenesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Autophagosome formation is initiated by a precursor isolation membrane (phagophore), to which two ubiquitin-like conjugation complexes are recruited. One of these involves the ubiquitin-like autophagy-related protein Atg8 (also called LC3) that is conjugated with a membrane phospholipid, phosphatidylethanolamine (PE). LC3-PE mediates membrane tethering and expansion of the phagophore that associates with and encircles the intracellular pathogen or the pathogen-containing vesicle. The second ubiquitin-like complex is comprised of ubiquitin-like Atg12 conjugated with the adaptor protein Atg5 and noncovalently associated with the adaptor protein Atg16. The Atg12-Atg5-Atg16 complex oligomerizes and is recruited to the newly formed isolation membrane and controls elongation and eventual closure of the phagophore.
Intracellular pathogens can be targeted for degradation through autophagy ( Figure 3-10). Bacteria that escape into the cytosol of the cell become ubiquitinated on the surface, which recruits adaptor proteins, such as NDP52 or p62, which can bind both ubiquitinated proteins on the bacterial surface and LC3-PE attached to phagophores. Another adaptor protein is optineurin (OPTN), which binds LC3 and ubiquitinated bacteria when it becomes phosphorylated by the kinase TBK1, a regulator of NF-κB signaling and interferon production.
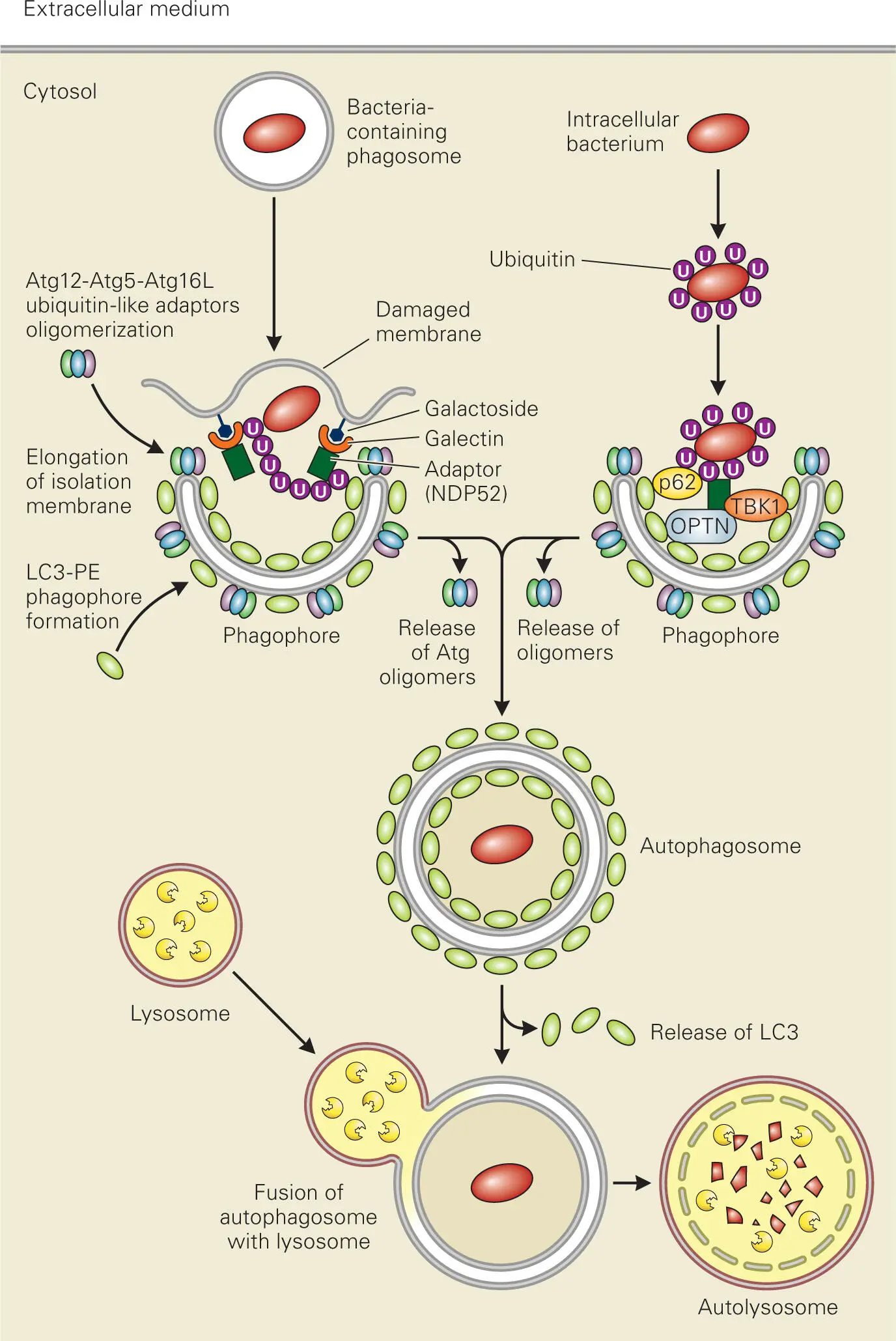
Figure 3-10. Eliminating intracellular pathogens through autophagy. Intracellular pathogens that escape into the cytosol or invading pathogens that enter via phagosomes or reside in pathogen-specific vacuoles are targeted for lysosomal degradation by autophagy. The target cytosolic pathogen or pathogen-containing vesicle is engulfed into a specialized double-membrane organelle called an autophagosome, which then fuses with a lysosome to become an autolysosome.
In the case of phagosomes containing invasive bacteria, such as M. tuberculosis or S. Typhimurium, autophagy is triggered by the damage to the phagosomal membrane caused by the bacterium. Recently, galectin-8 (a β-galactoside-binding lectin) has been identified as a cytosolic detector protein that can initiate the autophagy response for invading pathogens. Galectin-8 recognizes galactoside residues in exposed, damaged membrane and recruits autophagy adaptor proteins, such as NDP52, which binds to the autophagy proteins LC3 and Atg12 and brings the phagophore to the altered bacterium-containing phagosome. NOD2 protein can also initiate autophagy through recruitment and activation of the autophagy protein Atg16, which controls elongation of new autophagosomal membrane, at the site of bacterial entry into the cell.
The Complement Cascade
Complement Proteins
The skin and mucosa barriers, along with the phagocytic cells, are powerful defenses against bacterial invaders. There is, however, another highly complex and equally important arm of the innate defenses—the blood proteins that mark targets for phagocytic destruction and direct the inflammatory response. These complement system proteins are produced by the liver, circulate in the blood, and enter tissues throughout the body. Their primary purpose is to help phagocytic cells and antibodies rapidly clear pathogens from the body. Along with chemokines and cytokines, complement proteins also form an important link between the innate phagocytic defense system and the adaptive immune defenses, which will be discussed further in chapter 4.
Many of the complement proteins are zymogens, which reside throughout the body as inactive precursors. Complement activation occurs during infection as the complement proteins undergo a cascade of activating proteolytic cleavages, where each activated complement zymogen cleaves and activates the next complement zymogen. In terms of nomenclature, a letter “C” is used to designate the core complement components, some of which are actually multi-protein complexes. There are nine of these core components, C1 to C9. Activated proteolytic cleavage products are indicated by an “a” or “b.” In most cases, as, for example, C3, C4, and C5, “a” designates the smaller and “b” the larger of the two proteolytic products (e.g., C3a and C3b, respectively), but this rule is not followed uniformly, notably with regard to the complement component C2. Unfortunately for us, most immunologists prefer to retain the old nomenclature for C2, where the small C2 cleavage product is designated C2b and the large one C2a. We will use this designation.
Overview of Complement Pathways and Their Function
When activated through proteolytic cleavage and complex formation, the complement system performs five important interrelated functions: (1) promotion of opsonization (engulfment) of invading bacteria by coating them with complement components (e.g., C3b); (2) enhancement of chemotaxis of phagocytes (PMNs, monocytes, macrophages, and DCs) to attract them to the site of infection by releasing chemokines (e.g., C5a); (3) enhancement of vascular permeability to promote exudation (transmigration) of phagocytes from blood vessels into tissues at the site of infection by releasing vasodilators (e.g., C3a, C4a, and C5a) that cause degranulation (release of histamine and heparin) of basophils and mast cells; (4) promotion of agglutination (clustering and clumping) of invading bacteria by binding them together and increasing the efficiency of phagocytosis and clearance from the system; and (5) direct killing of many Gram-negative bacteria by poking holes in their membranes via binding of C5b to LPS and formation of the membrane attack complex (MAC) that consists of components C5b through C9.
Complement activation can be initiated through three pathways, as shown in Figure 3-11. The first two pathways are antibody-independent and form part of the innate response to initial infection, while the third pathway is antibody-dependent and comes into play after adaptive immunity has been launched. All three pathways lead to activation of the pivotal complement component, the protease C3-convertase, that cleaves and activates C3 into C3a and C3b. C3b covalently binds to the surface of pathogens, coating them and enhancing agglutination and opsonization of the pathogen by phagocytes.
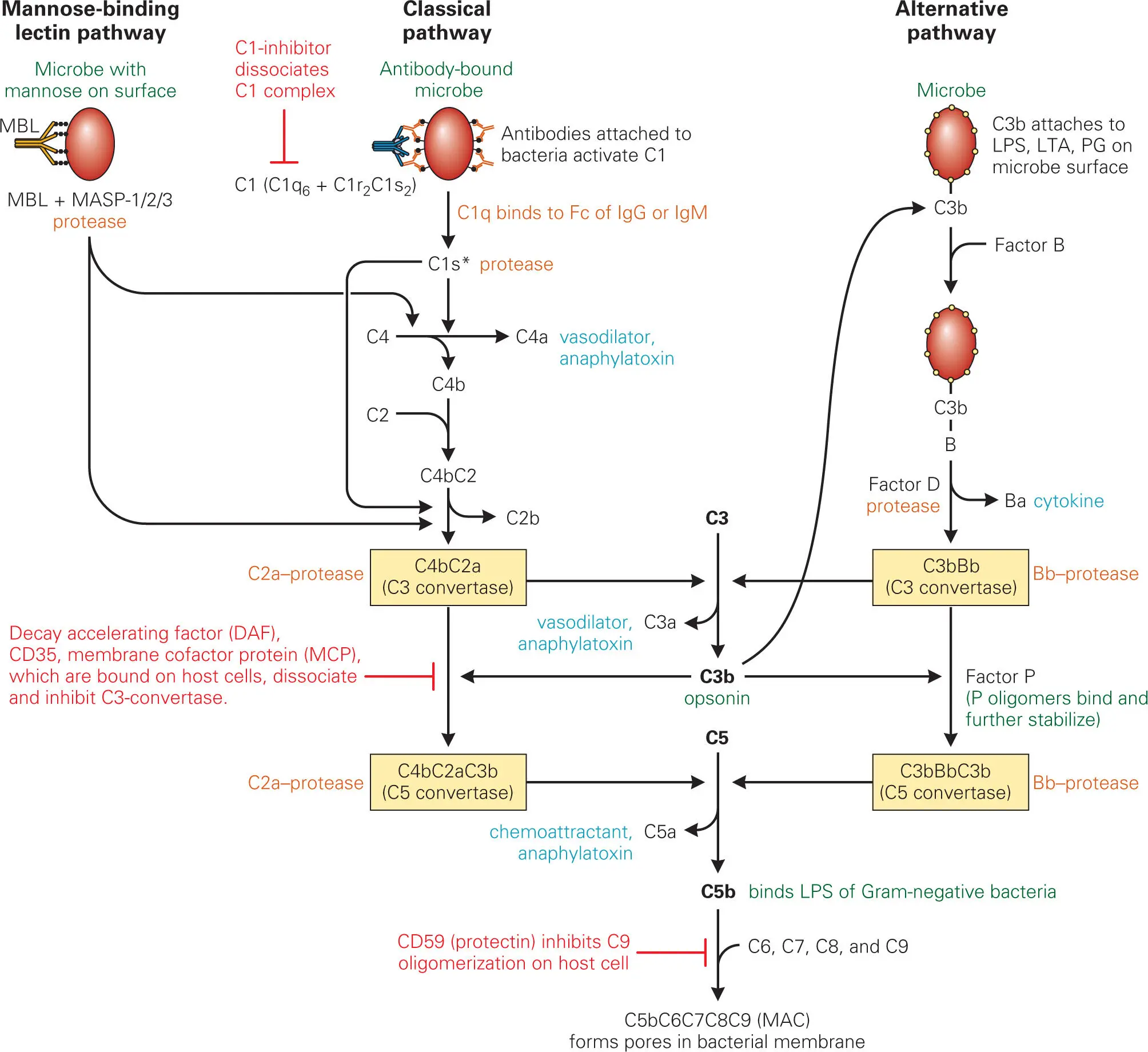
Figure 3-11. Main steps in activation of complement by the mannose-binding lectin, classical, and alternative pathways. These pathways differ only in the steps that initiate formation of C3 convertase. Important activated products are C3b (which opsonizes bacteria), C3a (which acts as a vasodilator), C5a (which acts as a vasodilator and a chemokine that attracts phagocytes to the area), and C5b-C9 (MAC [membrane attack complex], which inactivates enveloped viruses and kills Gram-negative bacteria). LPS, lipopolysaccharide.
Certain surface components of bacteria can trigger the complement cascade through the alternative pathway without the need for adaptive responses. The best-characterized complement-triggering bacterial surface molecules are LPS and LTA found in the outer and inner cell membranes of Gram-negative and Gram-positive bacteria, respectively. Complement-activating molecules of fungi, protozoa, and metazoa are not as well characterized, but they also appear to be lipid-carbohydrate complexes on the microbial cell surface. In the alternative pathway, the free hydroxyl (−OH) groups or amino (−NH2) groups of bacterial surface components react with an activated thioester group that is present in the C3b protein to form a covalent linkage that triggers the alternative pathway by recruiting factor B to the bacterial surface and initiating formation of the C3-convertase.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Bacterial Pathogenesis»
Представляем Вашему вниманию похожие книги на «Bacterial Pathogenesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Bacterial Pathogenesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.