Bioinformatics
Здесь есть возможность читать онлайн «Bioinformatics» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Bioinformatics
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Bioinformatics: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Bioinformatics»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
“This book is a gem to read and use in practice.”
— "This volume has a distinctive, special value as it offers an unrivalled level of details and unique expert insights from the leading computational biologists, including the very creators of popular bioinformatics tools."
— “A valuable survey of this fascinating field. . . I found it to be the most useful book on bioinformatics that I have seen and recommend it very highly.”
— “This should be on the bookshelf of every molecular biologist.”
— The field of bioinformatics is advancing at a remarkable rate. With the development of new analytical techniques that make use of the latest advances in machine learning and data science, today’s biologists are gaining fantastic new insights into the natural world’s most complex systems. These rapidly progressing innovations can, however, be difficult to keep pace with.
The expanded fourth edition of the best-selling
aims to remedy this by providing students and professionals alike with a comprehensive survey of the current field. Revised to reflect recent advances in computational biology, it offers practical instruction on the gathering, analysis, and interpretation of data, as well as explanations of the most powerful algorithms presently used for biological discovery.
offers the most readable, up-to-date, and thorough introduction to the field for biologists at all levels, covering both key concepts that have stood the test of time and the new and important developments driving this fast-moving discipline forwards.
This new edition features:
New chapters on metabolomics, population genetics, metagenomics and microbial community analysis, and translational bioinformatics A thorough treatment of statistical methods as applied to biological data Special topic boxes and appendices highlighting experimental strategies and advanced concepts Annotated reference lists, comprehensive lists of relevant web resources, and an extensive glossary of commonly used terms in bioinformatics, genomics, and proteomics
is an indispensable companion for researchers, instructors, and students of all levels in molecular biology and computational biology, as well as investigators involved in genomics, clinical research, proteomics, and related fields.
Bioinformatics — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Bioinformatics», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
BLAST 2 Sequences
A variation of BLAST called BLAST 2 Sequences can be used to find local alignments between any two protein or nucleotide sequences of interest (Tatusova and Madden 1999). Although the BLAST engine is used to find the best local alignment between the two sequences, no database search is performed. Rather, the two sequences to be compared are specified in advance by the user. The method is particularly useful for comparing sequences that have been determined to be homologous through experimental methods or for making comparisons between sequences from different species. Returning to the Protein BLAST (BLASTP) search page shown in Figure 3.6, checking the box marked “Align two or more sequences” will change the structure of the page, now allowing for the user to enter both the query and subject sequences that will be compared with one another ( Figure 3.11). As with any BLAST search, the user can adjust the standard array of BLAST-related options, including the selection of scoring matrix and gap penalties. A sample of the results produced by the BLAST 2 Sequences method is shown in Figure 3.12, comparing the transcription factor SOX-1 from H . sapiens and the ctenophore Mnemiopsis leidyi , the earliest branching animal species dating back at least 500 million years in evolutionary time (Ryan et al. 2013; Schnitzler et al. 2014). The major difference between this output and the typical BLAST output is the inclusion of a dot matrix view of the alignment, or “dotplot.” Dotplots are intended to provide a graphical representation of the degree of similarity between the two sequences being compared, allowing for the quick identification of regions of local alignment, direct or inverted repeats, insertions, deletions, and low-complexity regions. The dotplot in Figure 3.12indicates two regions of alignment, and additional information on those two regions of alignment is provided in the Alignments section at the bottom of the figure. As with all BLAST searches, the Alignments section provides the user with the usual set of scores, the E value, and percentages for identities, positives, and any gaps that may have been introduced.
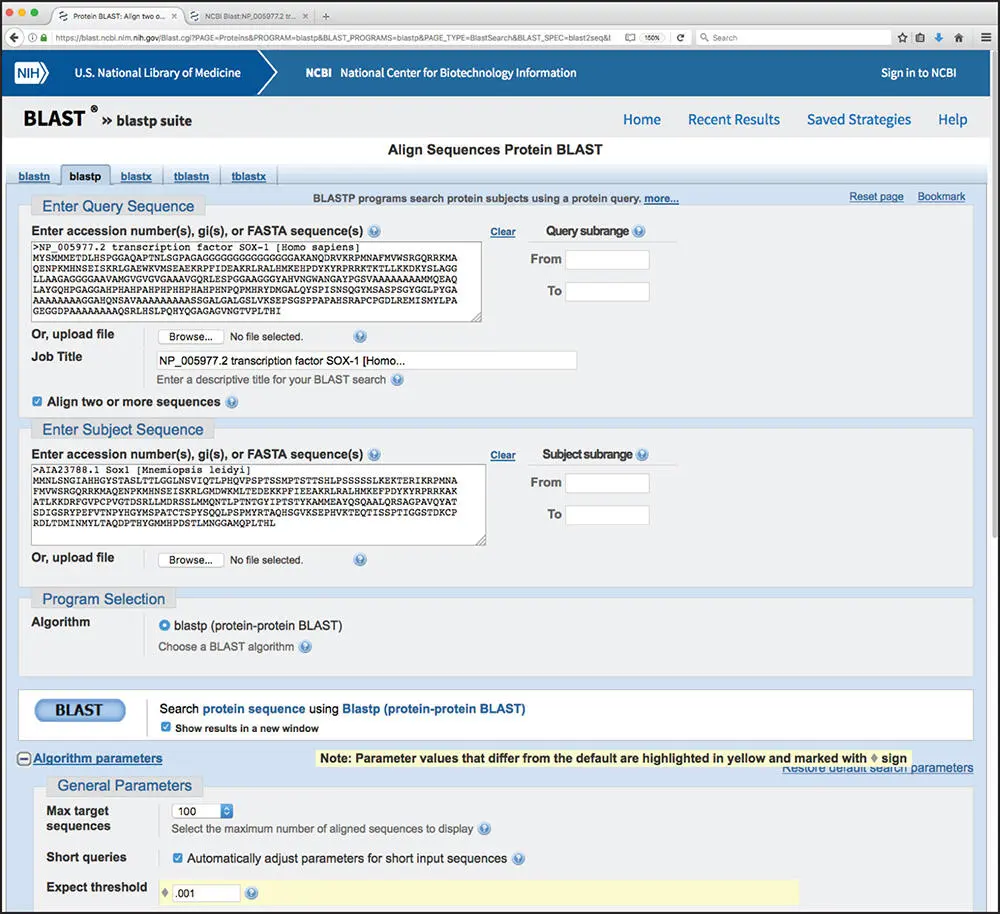
Figure 3.11 Performing a BLAST 2 Sequences alignment. Clicking the check box at the bottom of the Enter Query Sequence section expands the search page, generating a new Enter Subject Sequence section. Here, sequences for the transcription factor SOX-1 from human and the ctenophore Mnemiopsis leidyi have been used as the query and subject, respectively (Schnitzler et al. 2014). Here, only the BLASTP algorithm is available in the Program Selection section, as a one-to-one alignment has already been specified. The usual set of algorithm parameters is available, allowing the user to fine-tune the alignment as needed.
MegaBLAST
MegaBLAST is a variation of the BLASTN algorithm that has been optimized specifically for use in aligning either long or highly similar (>95%) nucleotide sequences and is a method of choice when looking for exact matches in nucleotide databases. The use of a greedy gapped alignment routine (Zhang et al. 2000) allows MegaBLAST to handle longer nucleotide sequences approximately 10 times faster than BLASTN would. MegaBLAST is particularly well suited to finding whether a sequence is part of a larger contig, detecting potential sequencing errors, and for comparing large, similar datasets against each other. The run speeds that are achieved using MegaBLAST come from changing two aspects of the traditional BLASTN routine. First, longer default word lengths are used; in BLASTN, the default word length is 11, whereas MegaBLAST uses a default word length of 28. Second, MegaBLAST uses a non-affine gap penalty scheme, meaning that there is no penalty for opening the gap; there is only a penalty for extending the gap, with a constant charge for each position in the gap. MegaBLAST is capable of accepting batch queries by simply pasting multiple sequences in FASTA format or a list of accession numbers into the query window.
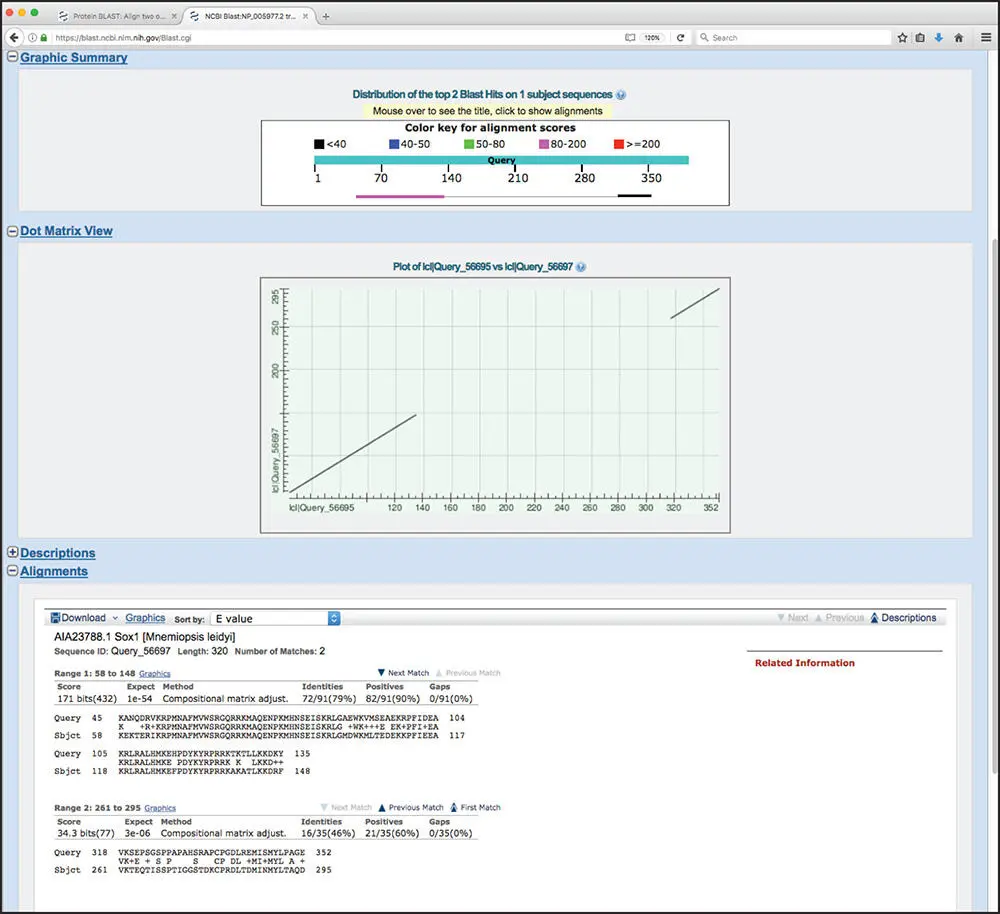
Figure 3.12 Typical output from a BLAST 2 Sequences alignment, based on the query issued in Figure 3.11. The standard graphical view is shown at the top of the figure, here indicating two high-scoring segment pairs (HSPs) for the alignment of the sequences for the transcription factor SOX-1 from human and the ctenophore Mnemiopsis leidyi . The dot matrix view is an alternative view of the alignment, with the query sequence represented on the horizontal axis and the subject sequence represented by the vertical axis; the diagonal indicates the regions of alignment captured within the two HSPs. The detailed alignments are shown at the bottom of the figure, along with the E values and alignment statistics for each HSP.
There is also a variation of MegaBLAST called discontiguous MegaBLAST. This version has been designed for comparing divergent sequences from different organisms, sequences where one would expect there to be low sequence identity. This method uses a discontiguous word approach that is quite different from those used by the rest of the programs in the BLAST suite. Here, rather than looking for query words of a certain length to seed the search, non-consecutive positions are examined over longer sequence segments (Ma et al. 2002). The approach has been shown to find statistically significant alignments even when the degree of similarity between sequences is very low.
PSI-BLAST
The variation of the BLAST algorithm known as PSI-BLAST (for position-specific iterated BLAST) is particularly well suited for identifying distantly related proteins – proteins that may not have been found using the traditional BLASTP method (Altschul et al. 1997; Altschul and Koonin 1998). PSI-BLAST relies on the use of position-specific scoring matrices (PSSMs), which are also often called hidden Markov models or profiles (Schneider et al. 1986; Gribskov et al. 1987; Staden 1988; Tatusov et al. 1994; Bücher et al. 1996). PSSMs are, quite simply, a numerical representation of a multiple sequence alignment, much like the multiple sequence alignments that will be discussed in Chapter 8. Embedded within a multiple sequence alignment is intrinsic sequence information that represents the common characteristics of that particular collection of sequences, frequently a protein family. By using a PSSM, one is able to use these embedded, common characteristics to find similarities between sequences with little or no absolute sequence identity, allowing for the identification and analysis of distantly related proteins. PSSMs are constructed by taking a multiple sequence alignment representing a protein family and then asking a series of questions, as follows.
What residues are seen at each position of the alignment?
How often does a particular residue appear at each position of the alignment?
Are there positions that show absolute conservation?
Can gaps be introduced anywhere in the alignment?
As soon as those questions are answered, the PSSM is constructed, and the numbers in the table now represent the multiple sequence alignment ( Figure 3.13). The numbers within the PSSM reflect the probability of any given amino acid occurring at each position. The PSSM numbers also reflect the effect of a conservative or non-conservative substitution at each position in the alignment, much like the PAM or BLOSUM matrices do. This PSSM now can be used for comparison against single sequences, or in an iterative approach where newly found sequences can be incorporated into the original PSSM to find additional sequences that may be of interest.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Bioinformatics»
Представляем Вашему вниманию похожие книги на «Bioinformatics» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.




Обсуждение, отзывы о книге «Bioinformatics» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.