Bioinformatics
Здесь есть возможность читать онлайн «Bioinformatics» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Bioinformatics
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Bioinformatics: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Bioinformatics»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
“This book is a gem to read and use in practice.”
— "This volume has a distinctive, special value as it offers an unrivalled level of details and unique expert insights from the leading computational biologists, including the very creators of popular bioinformatics tools."
— “A valuable survey of this fascinating field. . . I found it to be the most useful book on bioinformatics that I have seen and recommend it very highly.”
— “This should be on the bookshelf of every molecular biologist.”
— The field of bioinformatics is advancing at a remarkable rate. With the development of new analytical techniques that make use of the latest advances in machine learning and data science, today’s biologists are gaining fantastic new insights into the natural world’s most complex systems. These rapidly progressing innovations can, however, be difficult to keep pace with.
The expanded fourth edition of the best-selling
aims to remedy this by providing students and professionals alike with a comprehensive survey of the current field. Revised to reflect recent advances in computational biology, it offers practical instruction on the gathering, analysis, and interpretation of data, as well as explanations of the most powerful algorithms presently used for biological discovery.
offers the most readable, up-to-date, and thorough introduction to the field for biologists at all levels, covering both key concepts that have stood the test of time and the new and important developments driving this fast-moving discipline forwards.
This new edition features:
New chapters on metabolomics, population genetics, metagenomics and microbial community analysis, and translational bioinformatics A thorough treatment of statistical methods as applied to biological data Special topic boxes and appendices highlighting experimental strategies and advanced concepts Annotated reference lists, comprehensive lists of relevant web resources, and an extensive glossary of commonly used terms in bioinformatics, genomics, and proteomics
is an indispensable companion for researchers, instructors, and students of all levels in molecular biology and computational biology, as well as investigators involved in genomics, clinical research, proteomics, and related fields.
Bioinformatics — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Bioinformatics», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
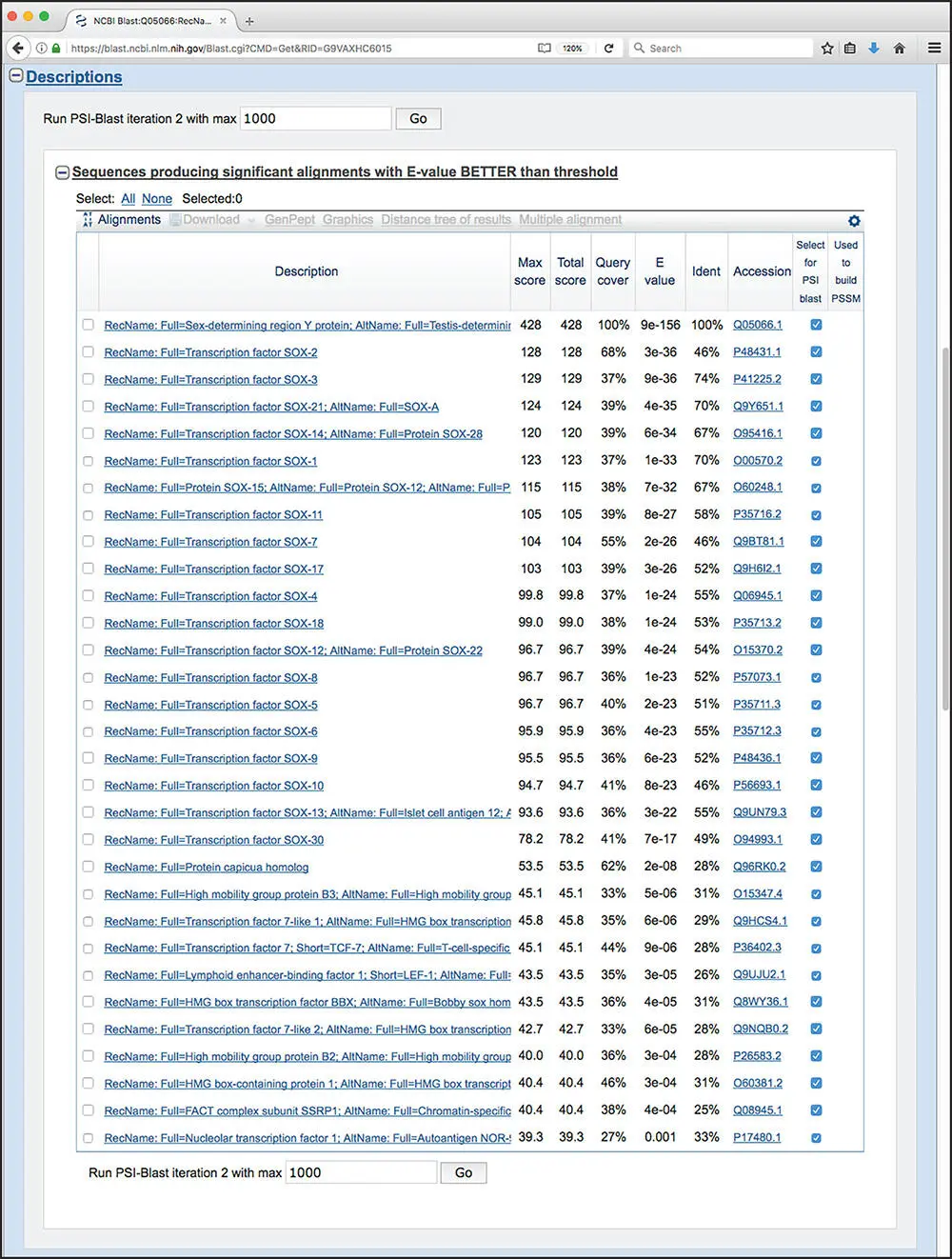
Figure 3.16 Results of the first round of a PSI-BLAST search. For each sequence found, the user is presented with the definition line from the corresponding UniProtKB/Swiss-Prot entry, the score value for the best high-scoring segment pair (HSP) alignment, the total of all scores across all HSP alignments, the percentage of the query covered by the HSPs, and the E value and percent identity for the best HSP alignment. The hyperlinked accession number allows for direct access to the source database record for that hit. Sequences whose “Select for PSI blast” box are checked will be used to calculate a position-specific scoring matrix (PSSM), and that PSSM then serves as the new “query” for the next round, the results of which are shown in Figure 3.17.
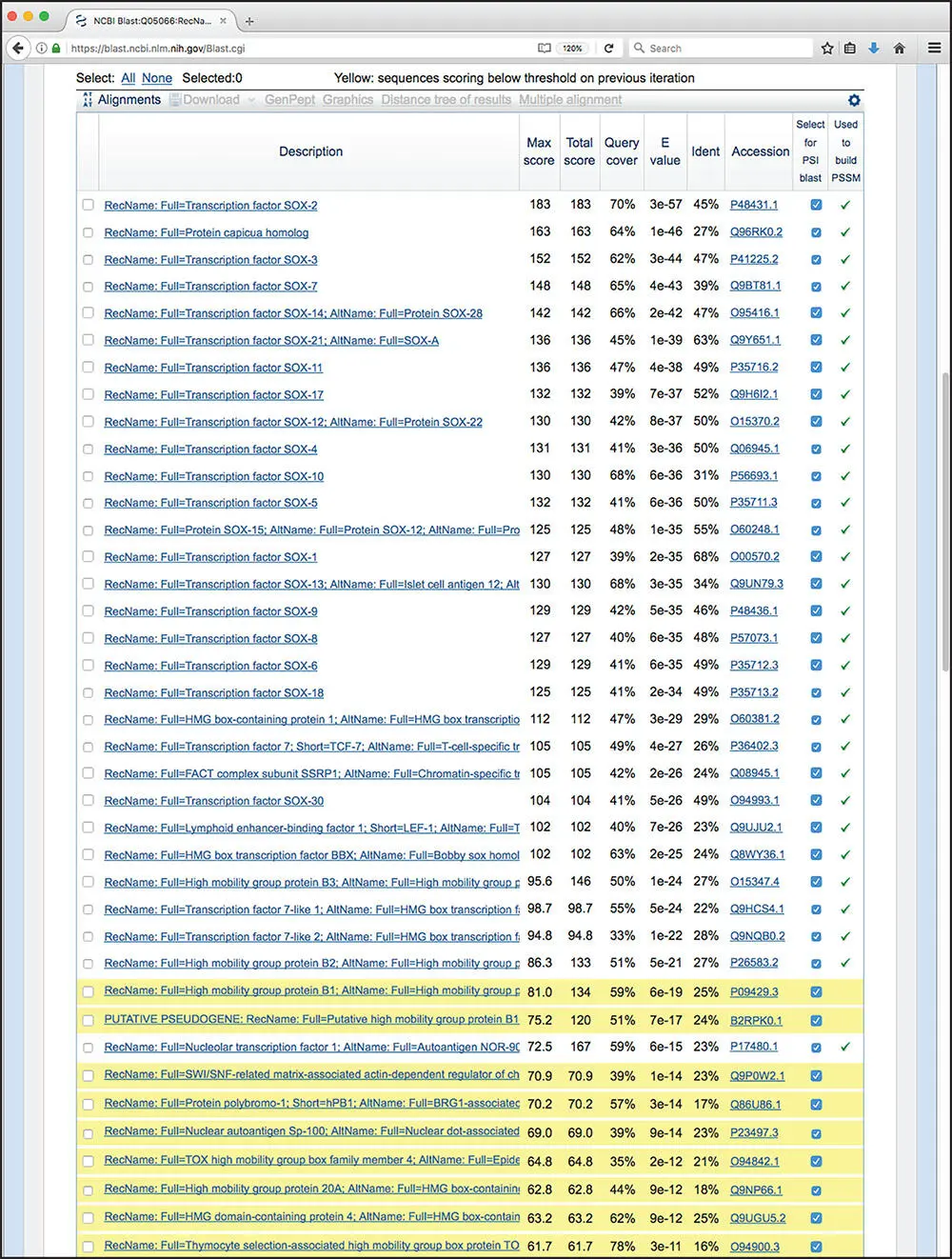
Figure 3.17 Results of the second round of a PSI-BLAST search. New sequences identified through the use of the position-specific scoring matrix (PSSM) calculated based on the results shown in Figure 3.16are highlighted in yellow. Check marks in the right-most column indicate which sequences were used to build the PSSM producing these results.
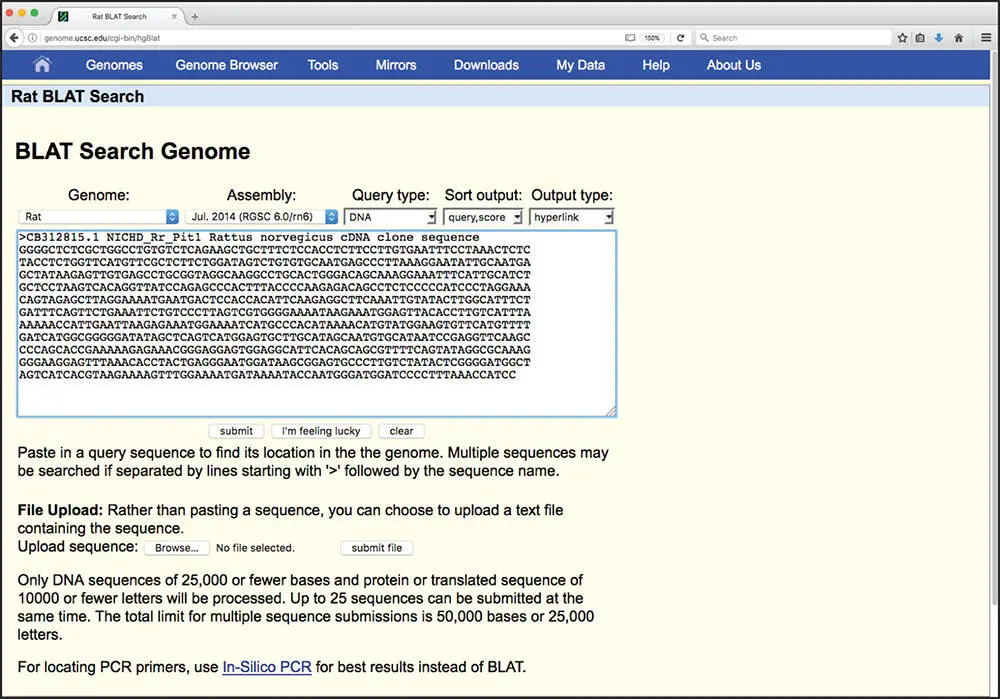
Figure 3.18 Submitting a BLAT query. A rat clone from the Cancer Genome Anatomy Project Tumor Gene Index (CB312815) is the query. The pull-down menus at the top of the page can be used to specify which genome should be searched (organism), which assembly should be used (usually, the most recent), and the query type (DNA, protein, translated DNA, or translated RNA). The “I'm feeling lucky” button returns only the highest scoring alignment and provides a direct path to the UCSC Genome Browser.
FASTA
While the most commonly used technique for detecting similarity between sequences is BLAST, it is not the only heuristic method that can be used to rapidly and accurately compare sequences with one another. In fact, the first widely used program designed for database similarity searching was FASTA (Lipman and Pearson 1985; Pearson and Lipman 1988; Pearson 2000). Like BLAST, FASTA enables the user to rapidly compare a query sequence against large databases, and various versions of the program are available ( Table 3.3). In addition to the main implementations, a variety of specialized FASTA versions are available, described in detail in Pearson (2016). An interesting historical note is that the FASTA format for representing nucleotide and protein sequences originated with the development of the FASTA algorithm.
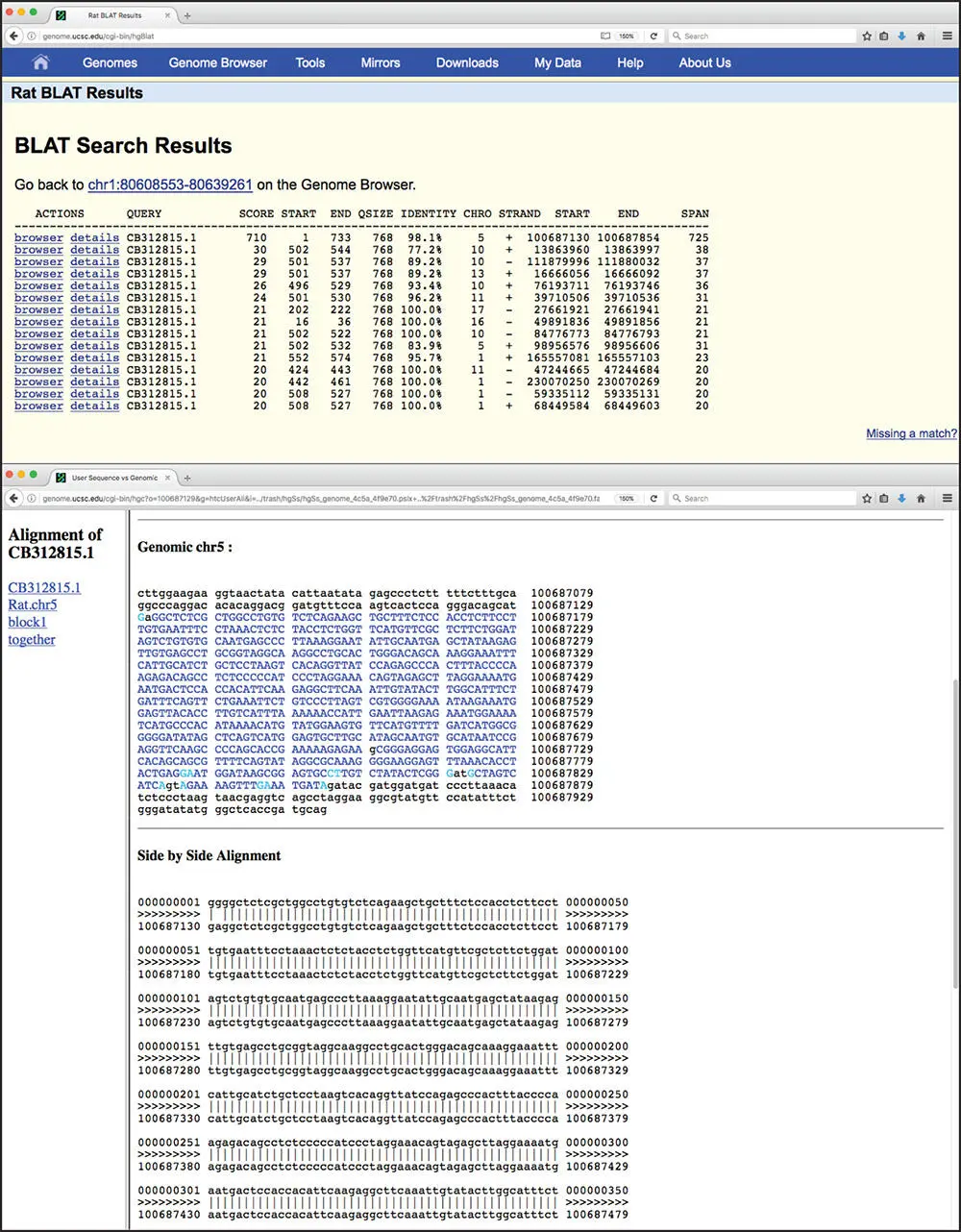
Figure 3.19 Results of a BLAT query. Based on the query submitted in Figure 3.18, the highest scoring hit is to a sequence on chromosome 5 rat genome having 98.1% sequence identity. Clicking on the “details” hyperlink brings the user to additional information on the found sequence, shown in the lower panel. Matching bases in the cDNA and genomic sequences are colored in dark blue and are capitalized. Lighter blue uppercase bases mark the boundaries of aligned regions and often signify splice sites. Gaps are indicated by lowercase black type. In the side-by-side alignment, exact matches are indicated by the vertical line between the sequences.
Table 3.3 Main FASTA algorithms.
| Program | Query | Database | Corresponding BLAST Program |
| FASTA | Nucleotide | Nucleotide | BLASTN |
| Protein | Protein | BLASTP | |
| FASTX/FASTY | DNA | Protein | BLASTX |
| TFASTYX/TFASTY | Protein | Translated DNA | TBLASTN |
The Method
The FASTA algorithm can be divided into four major steps. In the first step, FASTA determines all overlapping words of a certain length both in the query sequence and in each of the sequences in the target database, creating two lists in the process. Here, the word length parameter is called ktup , which is the equivalent of W in BLAST. These lists of overlapping words are compared with one another in order to identify any words that are common to the two lists. The method then looks for word matches that are in close proximity to one another and connects them to each other (intervening sequence included), without introducing any gaps. This can be represented using a dotplot format ( Figure 3.20a). Once this initial round of connections are made, an initial score ( init 1) is calculated for each of the regions of similarity.
In step 2, only the 10 best regions for a given pairwise alignment are considered for further analysis ( Figure 3.20b). FASTA now tries to join together regions of similarity that are close to each other in the dotplot but that do not lie on the same diagonal, with the goal of extending the overall length of the alignment ( Figure 3.20c). This means that insertions and deletions are now allowed, but there is a joining penalty for each of the diagonals that are connected. The net score for any two diagonals that have been connected is the sum of the score of the original diagonals, less the joining penalty. This new score is referred to as init n.
In step 3, FASTA ranks all of the resulting diagonals, and then further considers only the “best” diagonals in the list. For each of the best diagonals, FASTA uses a modification of the Smith–Waterman algorithm (1981) to come up with the optimal pairwise alignment between the two sequences being considered. A final, optimal score ( opt ) is calculated on this pairwise alignment.
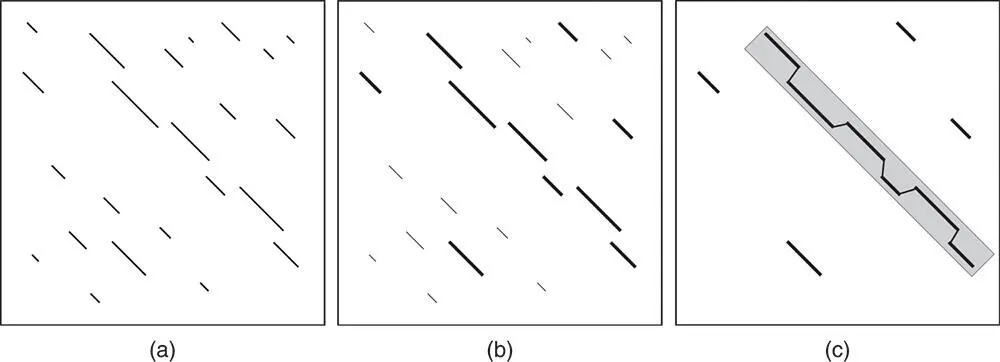
Figure 3.20 The FASTA search strategy. (a) Once FASTA determines words of length ktup common to the query sequence and the target sequence, it connects words that are close to each other, and these are represented by the diagonals. (b) After an initial round of scoring, the top 10 diagonals are selected for further analysis. (c) The Smith–Waterman algorithm is applied to yield the optimal pairwise alignment between the two sequences being considered. See text for details.
In the fourth and final step, FASTA assesses the significance of the alignments by estimating what the anticipated distribution of scores would be for randomly generated sequences having the same overall composition (i.e. sequence length and distribution of amino acids or nucleotides). Based on this randomization procedure and on the results from the original query, FASTA calculates an expectation value E (similar to the BLAST E value), which, as before, represents the probability that a reported hit has occurred purely by chance.
Running a FASTA Search
The University of Virginia provides a web front-end for issuing FASTA queries. Various protein and nucleotide databases are available, and up to two databases can be selected for use in a single run. From this page, the user can also specify the scoring matrix to be used, gap and extension penalties, and the value for ktup . The default values for ktup are 2 for protein-based searches and 6 for nucleotide-based searches; lowering the value of ktup increases the sensitivity of the run, at the expense of speed. The user can also limit the results returned to particular E values.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Bioinformatics»
Представляем Вашему вниманию похожие книги на «Bioinformatics» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.




Обсуждение, отзывы о книге «Bioinformatics» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.