Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
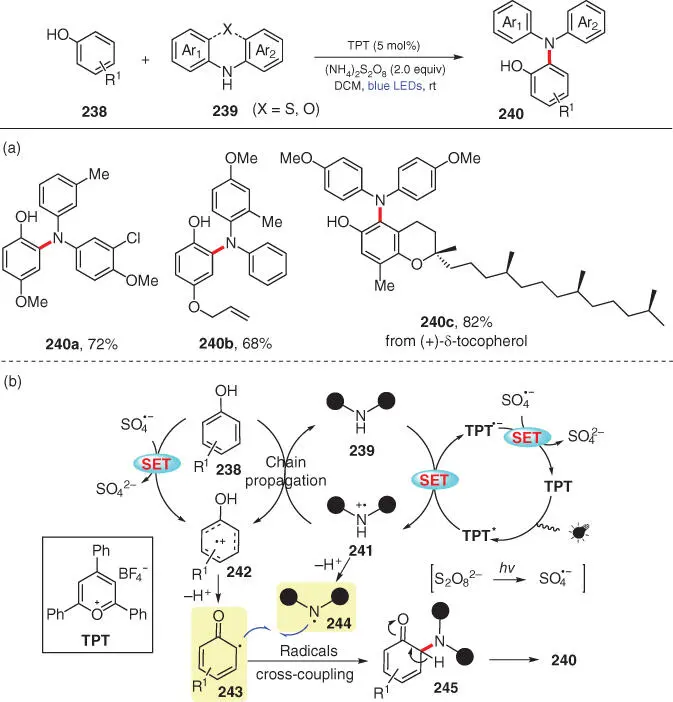
Scheme 3.41 Visible‐light‐mediated CDC amination of phenols and acyclic diarylamines.
Source: Modified from Zhao et al. [54].
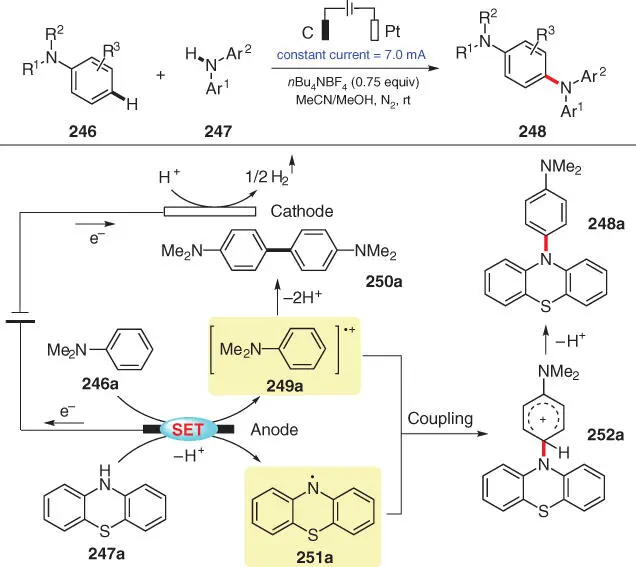
Scheme 3.42 Electro‐oxidative para‐selective C–H/N–H cross‐coupling with hydrogen evolution.
Source: Modified from Liu et al. [55].
An electrochemical protocol via a similar mechanism was described by Lei's group in early 2019, through which various triarylamines can be synthesized by coupling electron‐rich arenes and diarylamine derivatives ( Scheme 3.42) [55]. More specifically, a series of para‐selective amination products 248are furnished from the reactions between aniline derivatives 246and diarylamines 247. Both coupling partners are found to be redox active during the mechanistic investigation, as exemplified by a plausible mechanism related to 248a. Therefore, 246aand 247aare simultaneously oxidized at the anode to afford radical cation 249aand radical 251aafter deprotonation. The subsequent radical/radical cross‐coupling produces cationic species 252a, which turns into the final product 248aafter further deprotonation. Shortly after the publication of this work, Song and Li and coworkers also disclosed their independent research, which is very close to Lei's work [56].
3.4.1.2 Aryl C(sp 2)—N Bond Formation Using Azoles
In 2017, Adimurthy's group developed a regioselective C–H amination of heteroarenes with azoles using acridinium salt Mes‐Acr +ClO 4 −as the photocatalyst under visible light irradiation ( Scheme 3.43) [57]. Quinoline amides 253and imidazopyridines 254serve as eligible substrates, which undergo regioselective amination at C5 and C3 position, respectively, with various azoles 255. Taking the amination of 253as an example, it is proposed that the transformation proceeds through cross‐coupling of N‐radical cation 258and C‐radical species 260on the basis of a series of mechanistic investigations.
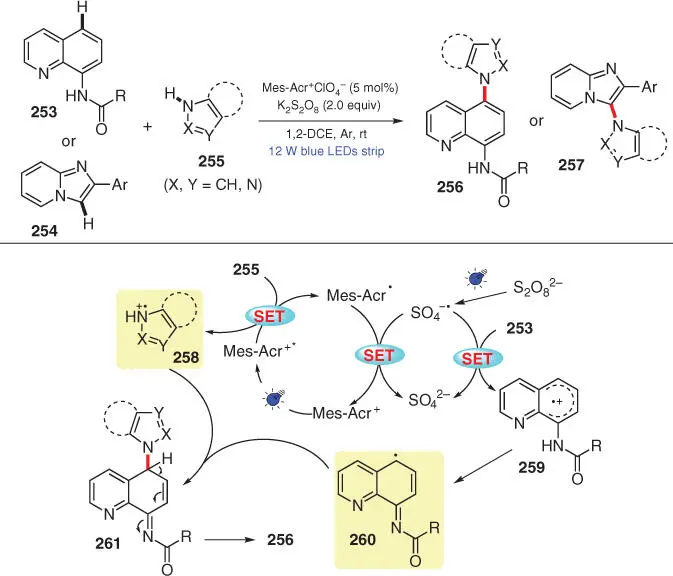
Scheme 3.43 Selective C–H amination of heteroarenes with azoles via an organic photoredox system.
Source: Modified from Samanta et al. [57].
More recently, Lei and coworkers disclosed an electrochemical C‐3 amination of imidazo[1,2‐ a ]pyridines 262with amines 263via an oxidative C–H/N–H cross‐coupling ( Scheme 3.44) [58]. The reaction is supposed to proceed via a cross‐coupling process of electrochemically generated C‐ and N‐radicals ( Scheme 3.44b). Additionally, an intramolecular C–N coupling through a Cp 2Fe‐mediated sequence of N‐radical formation and addition to the adjacent unsaturated moiety was also reported in the same article for the synthesis of 10 H ‐benzo[4,5]imidazo[1,2‐ a ]indole derivatives 266from 265.
One more electrochemical C–H azolation was next developed by Feng and Chen and coworkers in 2019, wherein phenol and aniline derivatives are regioselectively aminated with azoles via a radical–radical cross‐coupling pathway ( Scheme 3.45) [59]. During the investigation of the substrate scope, high ortho‐selectivity to the –XH group of this azolation is observed, and the sterically less‐hindered ortho‐position is preferred when asymmetrical phenols are applied ( 273b). In light of the mechanistic studies and reported work, the reaction mechanism is believed to involve a radical/radical cross‐coupling of 275and 276, which are generated from anodic oxidation of substrates 271and 272.
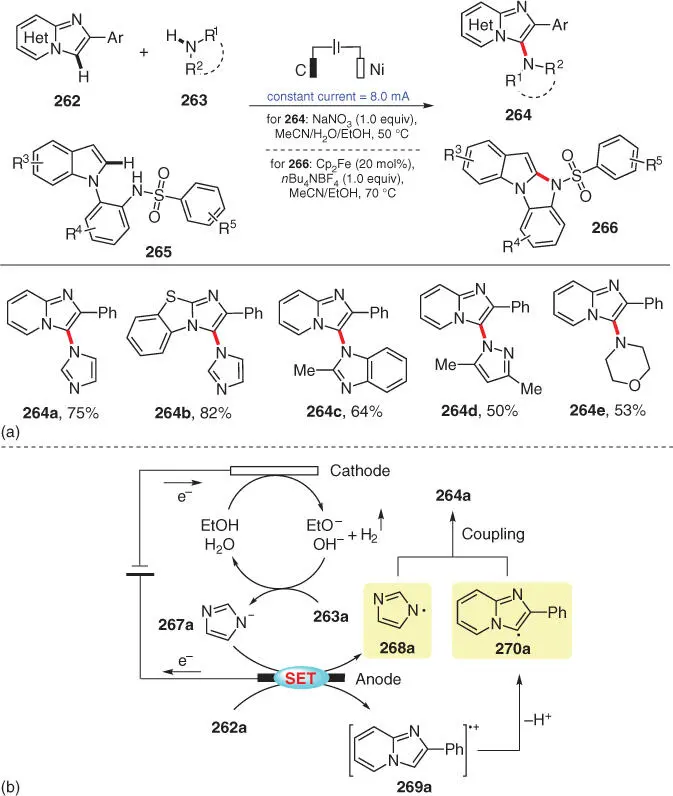
Scheme 3.44 Electrochemical oxidative C–H/N–H cross‐couplings for C—N bond formation with hydrogen evolution.
Source: Modified from Yu et al. [58].
3.4.2 Other C—N Bond Formation via Radical Cross‐coupling
The conjugate amination of α,β‐unsaturated carbonyl compounds represents a fundamental strategy to construct highly valuable β‐aminocarbonyl moieties. However, the direct addition of N‐centered radicals to electron‐deficient C=C bonds is unfavorable, at least as an intermolecular processes. To address this challenge, Meggers and coworkers realized a visible‐light‐induced enantioselective β‐amination of α,β‐unsaturated 2‐acyl imidazoles 278with N ‐aryl carbamates 279using an iridium‐based photocatalyst in combination with a chiral‐at‐rhodium Lewis acid (Δ‐RhO), through a PCET‐enabled N–H activation with the aid of a weak phosphate base, followed by a C–N radical cross‐coupling ( Scheme 3.46) [60]. The reaction mechanism is proposed in Scheme 3.46b, which begins with a PCET process between the photoexcited Ir III* and substrate 279with the aid of the Brønsted base, affording the carbamoyl radical 281and the reduced Ir IIspecies. On the other hand, chiral catalyst Δ‐RhO binds the α,β‐unsaturated compound 278to form the Rh‐bonded intermediate 282,which then undergoes single‐electron reduction with Ir IIto provide the Rh‐enolate radical intermediate 283and meanwhile regenerate photocatalyst Ir III. Subsequently, the cross‐coupling of radical intermediates 281and 283furnishes the C—N bond of 284. Alternative pathways via the addition of carbamate radical 281or the corresponding anion to the C=C bonds of Rh‐coordinated intermediate 282have been discussed and excluded on the basis of the experimental observations.
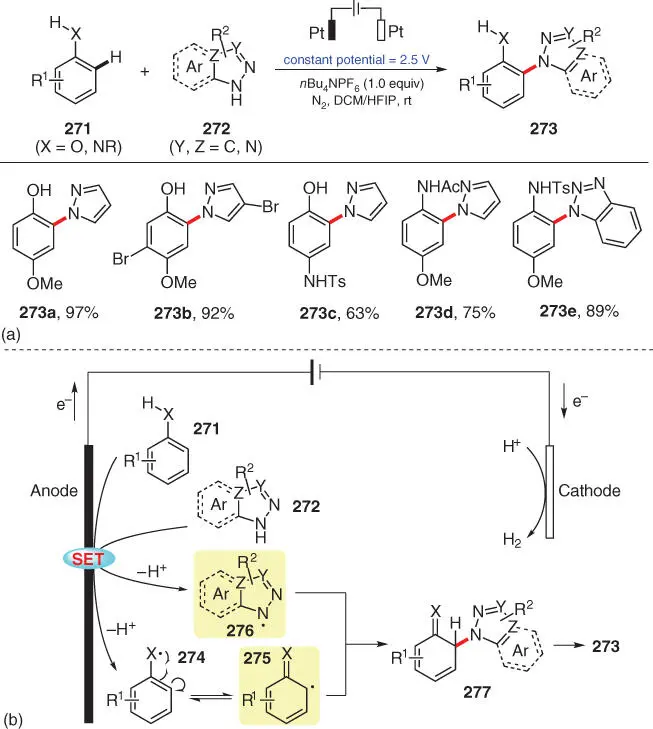
Scheme 3.45 Electro‐oxidative C–H azolation of phenol and aniline derivatives.
Source: Modified from Feng et al. [59].
Direct amination of C(sp 3)—H bonds via C/N‐radical cross‐coupling pathway often employs a HAT strategy (mainly 1,5‐HAT) and occurs intramolecularly. This topic has been discussed in detail within another chapter. As a distinct example, in 2017, Lei's group reported a metal‐ and oxidant‐free electrochemical protocol for the direct intermolecular C(sp 3)—N bond construction via a radical cross‐coupling pathway ( Scheme 3.47) [61]. The activated C(sp 3)—H bonds on benzylic or allylic sites, as well as those on the α‐positions to O, S, and N atoms ( 286), can be aminated with various azoles 287using this method ( 288a–288f). A tentative mechanism is proposed by the authors on the basis of the mechanistic studies and literature reports, as shown in Scheme 3.47b, in which N‐radical 289serves as both a HAT reagent to produce C‐radical 290and as a coupling partner to react with 290to afford C–N cross‐coupling product 288.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.