Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
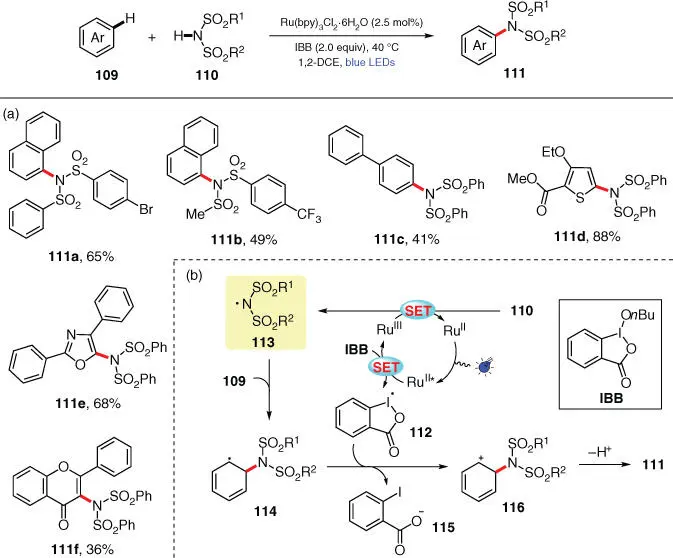
Scheme 3.20 Photocatalytic dehydrogenative C–H imidation of arenes with sulfonimides.
Source: Modified from Ito et al. [32].
Remarkably, aliphatic amines have also been successfully applied as aminating reagents for the CDC amination of arenes. In 2017, Nicewicz's group reported a challenging intermolecular C—N bond formation between simple arenes 117and primary aliphatic amines 118by means of photoredox catalysis ( Scheme 3.21) [33]. This transformation, which employs a mixed solvent of 1,2‐dichloroethane (1,2‐DCE) and phosphate buffer (pH = 8), accommodates a wide range of electron‐rich aromatic and heteroaromatic compounds and diverse primary aliphatic amines including amino acids. According to fluorescence quenching experiments, both the arenes and amines are able to quench the excited photocatalyst; thus, both of their radical cations are potential reaction intermediates. However, even the less electron‐rich arenes such as benzene and toluene, which are unlikely to be oxidized by the excited photocatalyst, can undergo smooth amination to afford the corresponding aniline products ( 119d, 119e). Consequently, an arene radical cation pathway can be excluded in such cases. Based on all mechanistic studies, a plausible mechanism is presented by the authors as shown in Scheme 3.21b. For the reactions of electron‐poor/neutral arenes, radical cation 120produced from SET oxidation of amine 118by the photoexcited Mes‐Acr +* is the only possible intermediate, which subsequently adds onto arene 117to form a cyclohexadienyl radical 121. Upon oxidation by O 2and deprotonation, 121aromatizes to form the desired amination product 119. As for the reactions of electron‐rich arenes, both amine radical cation and arene radical cation can serve as potential intermediates, and the mechanism via the latter will be discussed in Section 3.3.
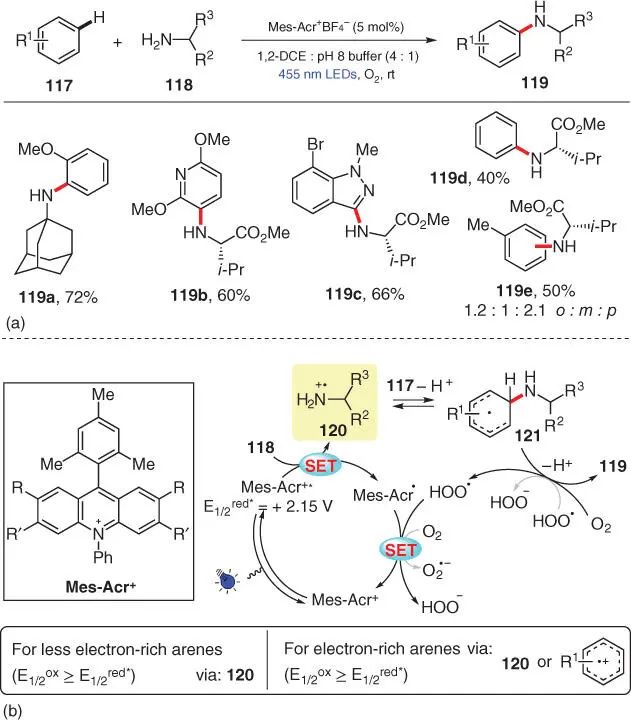
Scheme 3.21 Photocatalytic aryl C–H amination using primary aliphatic amines.
Source: Modified from Margrey et al. [33].
Organic electrochemistry has also been applied to generate N‐radicals directly from N—H bonds for addition to aromatic moieties. In 2017, Xu and coworkers described an electrochemical approach to access amidinyl radical 123via the anodic N—H bond cleavage of substrate 122( Scheme 3.22) [34]. Through the intramolecular cyclization of N‐radicals 123onto their arene and heteroarene moieties, various tetracyclic benzimidazoles or pyridoimidazoles 124are furnished with high efficiency. The reaction can also easily be scaled‐up by simply switching up the constant current ( 124a– 124d). Additionally, substrate 122ewith two potential cyclization sites is selected as an example to investigate the cyclization tendency of amidinyl radical 123e. Based on both experimental and theoretical results, 6‐endo‐trig cyclization (path a) to form a six‐membered ring takes precedence over the alternative five‐membered ring formation (path b).
In 2018, Lei's group developed a metal‐ and oxidant‐free electrochemical protocol for the oxidative C–H amination of unprotected phenols ( Scheme 3.23) [35]. Various phenothiazine derivatives 126generally couple at the ortho‐position of electron‐rich phenols 125, affording N ‐aryl phenothiazines 127in good to excellent yields. Low yields of 127are achieved using phenols with an electron‐withdrawing group ( 127b) or electron‐neutral diaryl amines ( 127g, 127h). When both ortho‐positions of the phenol are occupied, the amination occurs at the para‐position instead ( 127f). The reaction is believed to proceed via an N‐radical cation addition to the aromatic ring of the phenol ( Scheme 3.23b). Taking 127aas an example, phenothiazine 126ais first oxidized at the anode to generate a radical cation intermediate 128a, which then undergoes electrophilic addition to 125a, affording the hydroxyl carbon radical 129a. Subsequent anodic oxidation and deprotonation of 129agives the final C(sp 2)—N bond formation product 127a. Simultaneously, the reduction of protons at the cathode releases hydrogen gas during the process. In the following year, these authors have also applied the protocol to the modification of biomolecules [36].
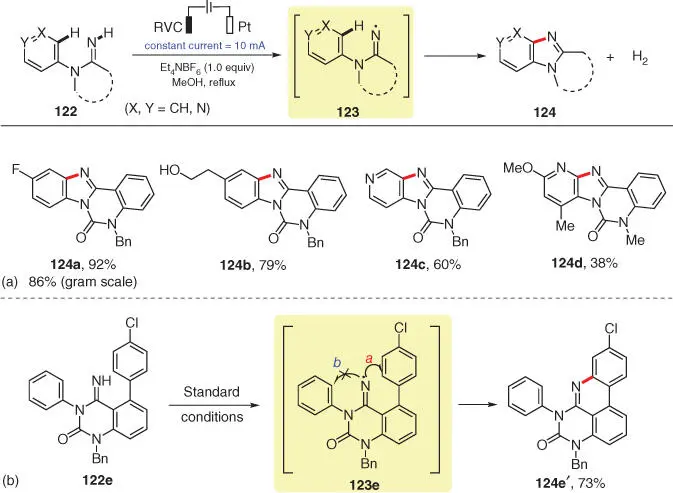
Scheme 3.22 Anodic N—H bond cleavage for aromatic C—H bond amination.
Source: Modified from Zhao et al. [34].
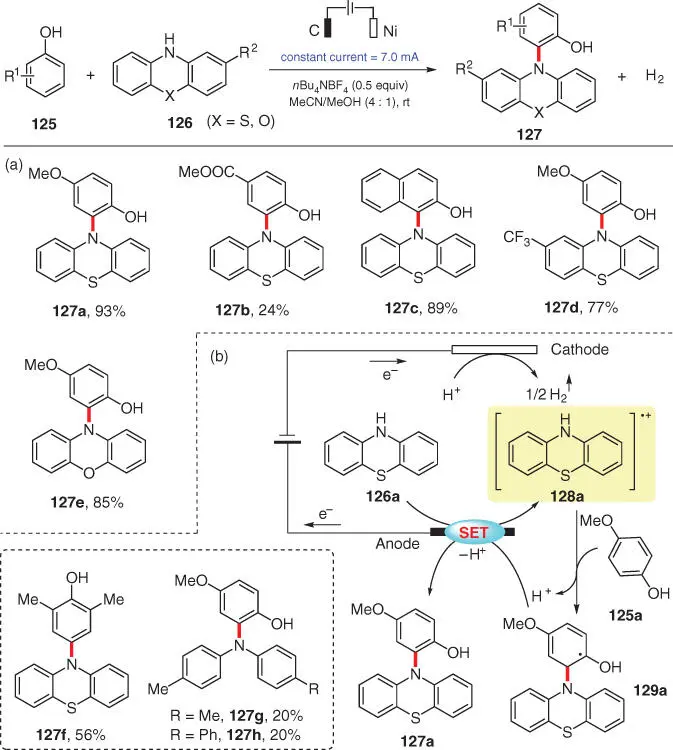
Scheme 3.23 Electrochemical oxidative C–H amination of phenols.
Source: Modified from Tang et al. [35].
3.3 Amination via N‐atom Nucleophilic Addition
As introduced in Section 3.2, construction of C—N bonds via N‐radical addition pathway relies on N‐centered radical (or radical cation) species, generated by oxidation of N—H bonds, as key intermediates. On the contrary, aminations via N‐atom nucleophilic addition pathway proceed through the oxidation of the C–H partners, and the amines usually serve as nucleophiles to attack the generated C‐centered cationic intermediates during the C—N bonds forming processes.
3.3.1 Aromatic C(sp 2)—H Bond Amination
As documented in the literature precedents, direct oxidation of (hetero)arenes is feasible under photo‐ or electrochemical conditions, and the resulting radical cation intermediates could then be attacked by nucleophiles such as –NHR, –OH, and –CN −. Yet, among the electrochemical amination reactions via arene radical cation intermediates, methods directly employing simple N–H nucleophiles are still not common. Nevertheless, notable progress has been advanced via photoredox catalysis, wherein strongly nucleophilic azoles usually serve as the amine partners.
In 2015, a photocatalytic site‐selective CDC amination of unfunctionalized arenes with azoles was described by Nicewicz's group using an organic catalyst system consisting of an acridinium photocatalyst and a nitroxyl radical ( Scheme 3.24) [37]. Upon irradiation with visible light, the photoexcited acridinium photocatalyst Mes‐Acr +* is able to directly oxidize arene 130into the corresponding radical cation 134, which is subsequently attacked by the amine nucleophile 131to form cationic radical 135. Deprotonation of 135affords radical intermediate 136, which further undergoes aromatization via a TEMPO‐mediated HAT process to furnish the final product 133. Alternatively, intermediate 136can also be trapped by O 2to form 1,3‐cyclohexadienyl peroxyl radical 137, which then converts into product 133after the internal elimination of the hydroperoxyl radical HOO ·. The closure of two catalytic cycles is indicated by the regeneration of ground‐state photocatalyst Mes‐Acr +from the SET oxidation of Mes‐Acr ·by O 2or HOO ·, and TEMPO from a HAT process between TEMPO‐H and O 2or O‐radical species such as O 2 ·−and HOO ·. In the scope exploration, a variety of aromatic compounds including complex drug‐like structures are firstly investigated, with the corresponding amination products obtained in moderate to good yields. A series of heterocyclic nucleophiles including pyrazoles, triazoles, tetrazoles, imidazoles, and benzimidazoles are proven as suitable coupling partners for the CDC amination with arenes. Particularly, reactions between a commercially available ammonium carbamate H 4N +H 2NCO 2 −( 132) and various (hetero)arenes directly furnish primary aniline products.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.