Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
In the typical visible‐light‐induced reactions described above, the photoredox catalysts harvest visible light to reach their excited states, which then undergo further single‐electron redox events with reactants. Recently, a new photoactivation paradigm has emerged, wherein the transition‐metal‐based photocatalysts also directly engage in the bond formation/cleavage processes. In this area, a growing number of photoinduced C—N bonds forming methodologies using Cu‐based unconventional photocatalysts have lately been developed by Fu, Hwang, Kobayashi, etc., and their progresses have been summarized by Gevorgyan in 2017 [28]. As an example, Hwang and coworkers reported a visible‐light‐induced, Cu‐catalyzed reaction for the preparation of α‐ketoamides from anilines 87and alkynes 88under ambient conditions ( Scheme 3.16) [29]. Using oxygen as a green oxidant, this ligand‐ and base‐free method features high atom economy and is compatible with a wide range of aniline and alkyne substrates. Notably, bioactive epoxide hydrolase inhibitors 89iand 89jare smoothly prepared within a single step from commercial starting materials by means of this strategy.
A plausible pathway is proposed on the basis of a series of detailed mechanistic studies ( Scheme 3.17). Via a weak Cu I–aniline complex formed by catalyst CuCl and aniline 87, Cu I–phenylacetylide 90is generated upon the addition of alkyne 88, which then reaches its excited state 91under blue light irradiation. The following SET oxidation of 91by O 2provides an electron‐deficient Cu II–phenylacetylide 92as well as a superoxide. The following nucleophilic addition of aniline 87to complex 92results in the Cu IIIspecies 93, which next undergoes reductive elimination to furnish the highly reactive Cu I‐coordinated ynamine 94. The intermediate 94readily reacts with O 2to generate the Cu IIperoxo‐complex 95, which then tautomerizes into the Cu Ispecies 96. Finally, complex 96is attacked by Cl −to regenerate the catalyst CuCl, and the subsequent ring cleavage of the resulting intermediate 97furnishes the final α‐ketoamide product 89.
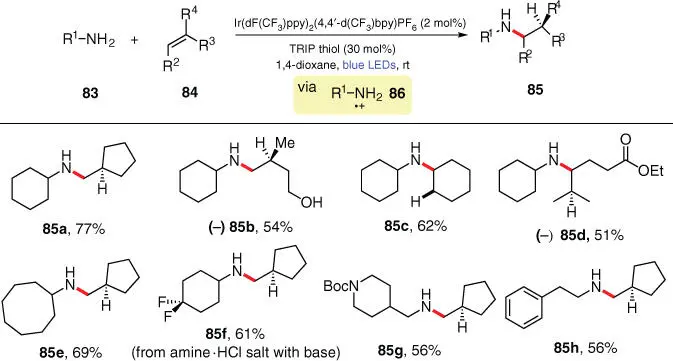
Scheme 3.15 Photocatalytic intermolecular anti‐Markovnikov hydroamination of unactivated olefins with primary alkyl amines.
Source: Miller et al. [27].
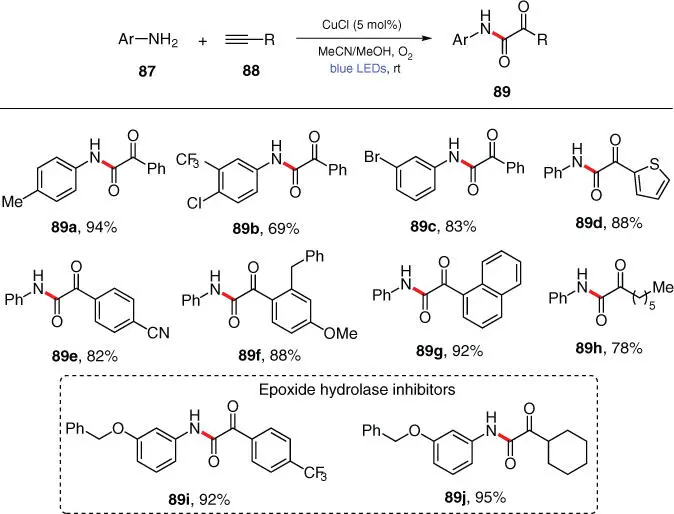
Scheme 3.16 Photoinduced, CuCl‐catalyzed oxidative C–N coupling of anilines with terminal alkynes.
Source: Modified from Sagadevan et al. [29].
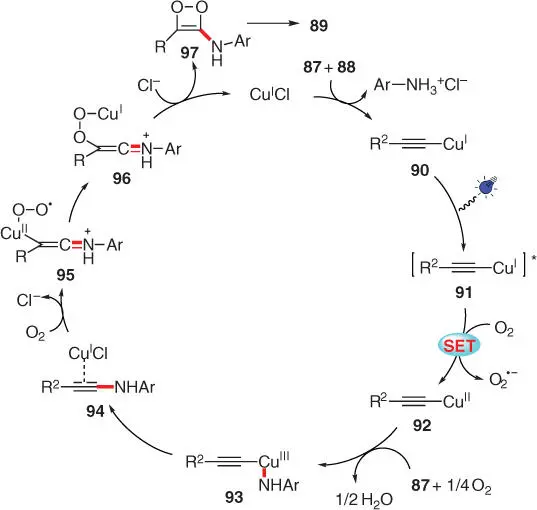
Scheme 3.17 Proposed mechanism for the photoinduced, CuCl‐catalyzed C–N coupling of anilines with terminal alkynes.
3.2.2 Radical Species Addition to Aromatic Rings
In the early years, the direct oxidative aminations of unfunctionalized arenes were achieved using iodine‐based reagents by the research groups of DeBoef, Chang, and Antonchick, wherein the employed amine partners were mainly sulfonamides and phthalimides. With the development of photoredox and electrochemical methodologies, more and more sustainable synthetic alternatives have emerged for the direct C(sp 2)—N bond formation of simple arenes via N‐radical species addition pathways.
In 2016, Yu and Zhang and coworkers reported a visible‐light‐induced CDC amination of heteroarenes 98with sulfonamides 99using bleach (aqueous NaClO solution) as the oxidant ( Scheme 3.18) [30]. Multiple heteroarenes such as indoles, pyrroles, and benzofurans can undergo this transformation with N ‐methyl‐ para ‐toluene sulfonamide to afford their corresponding C2‐amide products ( 100a– d). The scope of sulfonamides is also explored using 1,3‐dimethyl‐1 H ‐indole as the heteroarene partner, with R 1and R 2being aromatic or aliphatic moieties ( 100e– 100h). Furthermore, attempts to apply this protocol to intramolecular C2‐amination/cyclization of indoles succeeded in building fused indole derivatives ( 100i, 100j). The proposed mechanism is depicted in Scheme 3.18b. After excitation of the photocatalyst Ir IIIunder visible light irradiation, the excited Ir III* is then oxidized by NaClO to its higher valence state Ir IV, as demonstrated by fluorescence quenching experiments. The resulting Ir IVis capable of directly oxidizing sulfonamide 99into N‐centered radical 101, meanwhile regenerating Ir IIIto complete the photocatalytic cycle. The subsequent addition of N‐radical 101onto the C2 position of heteroarene 98forms benzylic radical 102, which undergoes further oxidation/deprotonation sequence to deliver the desired product 100via cationic intermediate 103.
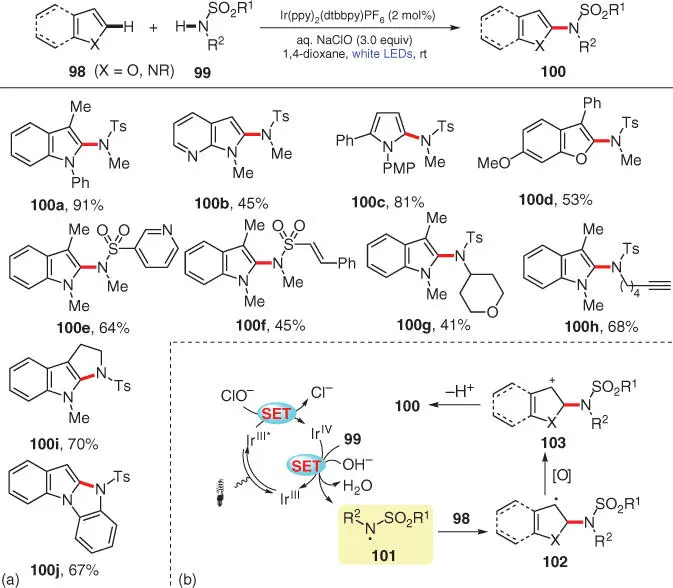
Scheme 3.18 Visible‐light‐induced direct C–H amination of heteroarenes with sulfonamides.
Source: Modified from Tong et al. [30].
In the following year, another CDC amination of heteroarenes 104using phthalimide 105as the N–H source was then disclosed by Itoh's group, employing 2‐ tert ‐butylanthraquinone (2‐ t ‐Bu‐AQN) as the photocatalyst and aerobic oxygen as the external oxidant ( Scheme 3.19) [31]. Multiple heteroarenes are smoothly aminated under the metal‐free conditions, including diverse N‐protected indoles and pyrroles as well as benzo[ b ]thiophene ( 106a–106d). Based on several control experiments, the authors postulate that, after the deprotonation by K 2CO 3and the single‐electron oxidation by the photoexcited AQN*, phthalimide 105is converted into the key N‐centered radical 107, and the resulting AQN ·−is then oxidized back to the original AQN by aerobic oxygen to complete the photocatalytic cycle. Subsequently, the desired amination product 106is furnished from intermediate 107after a radical addition/SET oxidation/aromatization sequence.
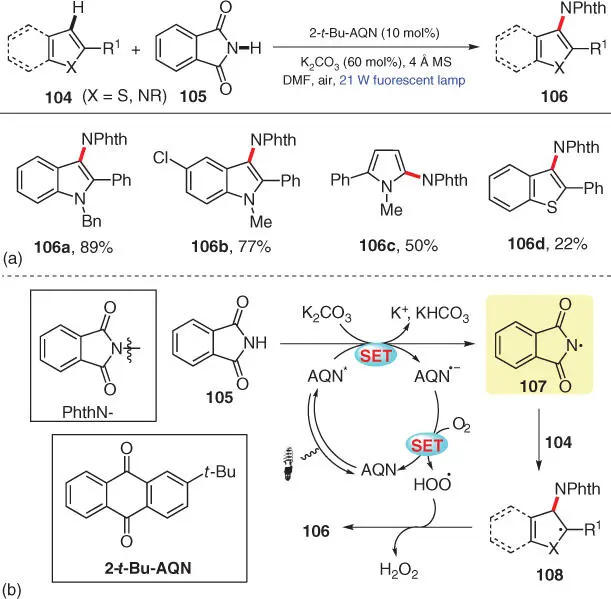
Scheme 3.19 Visible‐light‐mediated C–H amination of heteroarenes.
Source: Modified from Yamaguchi et al. [31].
In the same year, Itami and coworkers disclosed a CDC amination of aromatic C(sp 2)–H enabled by visible light photoredox catalysis, using sulfonimides 110as the N–H sources and a widely used Ru‐complex as the photocatalyst ( Scheme 3.20) [32]. The substrate scopes of both coupling partners are reasonable, covering a variety of arylsulfonimides bearing various substituents and different arenes and heterocycles ( 111a–111f). The mechanism proposed by the authors is presented in Scheme 3.20b. First, the hypervalent iodine oxidant 1‐butoxy‐1λ 3‐benzo[ d ][1,2]iodaoxol‐3(1 H )‐one (IBB) undergoes a SET reduction by the photoexcited catalyst Ru II* to generate a radical intermediate 112along with a Ru IIIspecies. The subsequent single‐electron oxidation of sulfonimide 110by Ru IIIyields the key imidyl radical 113, which then adds onto arene 109to afford radical intermediate 114. Next, cationic intermediate 116formed via a SET process between 112and 114, further aromatizes to afford the desired amination product 111.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.