Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
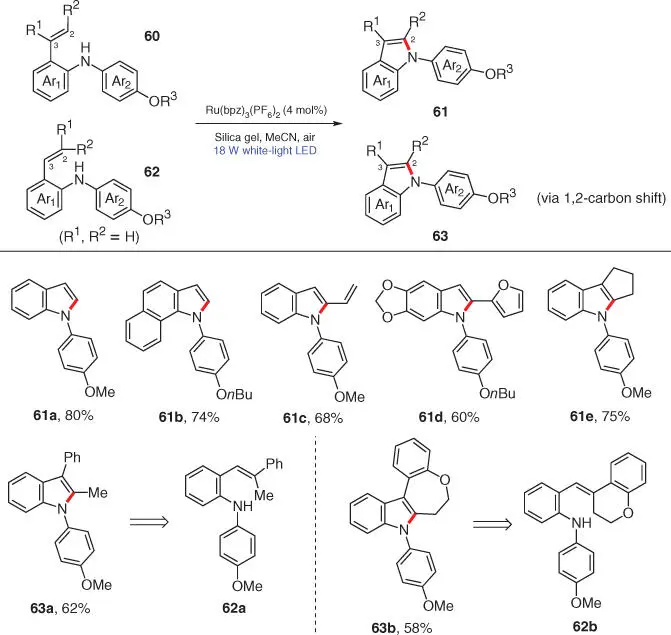
Scheme 3.10 Photocatalytic C—N bond formation for the preparation of N ‐arylindoles.
Source: Modified from Maity and Zheng [23].
A plausible mechanism for the above photocatalytic transformation is proposed in Scheme 3.11. The reaction occurs with the oxidation of substrate 60by the excited state photocatalyst Ru II* to generate the key aminium radical cation 64. The subsequent intramolecular electrophilic addition of 64to its tethered C=C bonds affords intermediate 65, which then turns into benzylic radical 66after a proton abstraction. The oxidation into the corresponding benzylic cation 67and the further aromatization of 67upon deprotonation finally result in the desired N ‐arylindole 61. As for substrate 62, the reaction proceeds through the same pathway before reaching benzylic cation 68. Then, a 1,2‐carbon shift event follows, in which one of the substituents on C2 migrates onto C3, providing cationic intermediate 69, which is further deprotonated to deliver the final product 63.
Thereafter, the authors applied this intramolecular C—N bond formation strategy and the same privileged structure to another photoredox cyclization protocol, in which two aliphatic rings are formed in one step to furnish fused indolines 71( Scheme 3.12) [24]. Herein, remote nucleophilic functional groups such as –OH and –NHBoc are installed at the C2 position of the 2‐styryl anilines 70to attack the in situ generated benzylic carbocations, and the desired cyclization products 71are successfully obtained under photocatalytic conditions similar to their previous work.
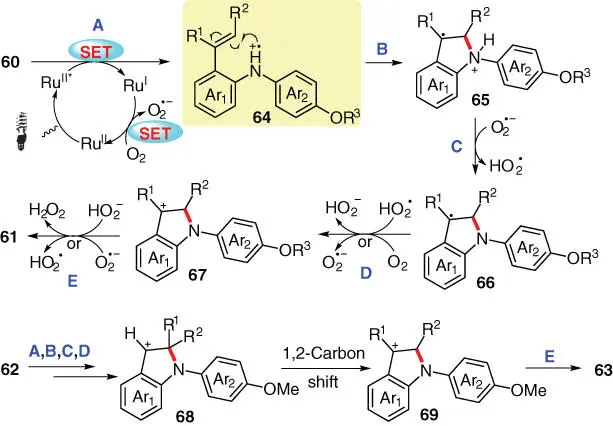
Scheme 3.11 Mechanistic proposal for the photocatalytic synthesis of N ‐arylindoles.
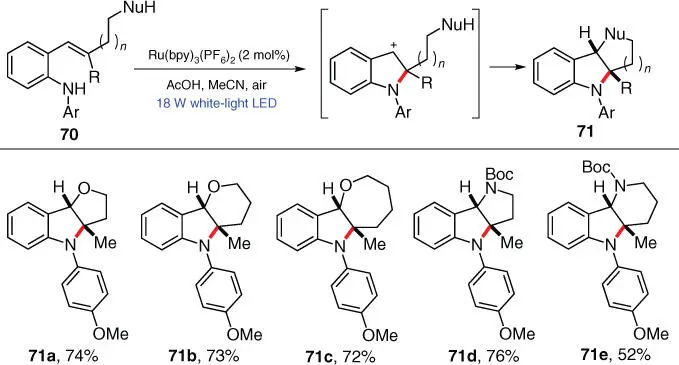
Scheme 3.12 Visible‐light‐initiated tandem reaction for the synthesis of fused N ‐arylindolines.
Source: Modified from Morris et al. [24].
In 2014, an intramolecular anti‐Markovnikov hydroamination protocol of aryl olefins was disclosed by Knowles' group, for the construction of structurally diverse N ‐aryl heterocycles 73in a simple reaction system under mild photocatalytic conditions ( Scheme 3.13) [25]. The reaction is proposed to begin with the direct oxidation of aniline 72by the photoexcited catalyst Ir III* to generate the crucial aminium radical cation 74, as supported by both CV data and luminescence quenching experiments. The following sequence of the intramolecular addition to the tethered alkene of intermediate 74, the one‐electron reduction of benzylic radical 75by Ir II, and the proton transfer from alcoholic solvent to 76delivers the final product 73. A wide range of substrates bearing various substituents on both aromatic groups (Ar 1and Ar 2) are proven compatible with this method, providing cyclization products in good to excellent yields, including five‐ or six‐membered, heterocylic, and bicyclic ones ( 73a–73h). However, application of this protocol to intermolecular couplings turned out to be unsuccessful, which is likely due to the fact that the bimolecular C—N bond formation fails to outcompete the favorable electron back‐transfer from the Ir IIcomplex to the aminium intermediate, as speculated by the authors.
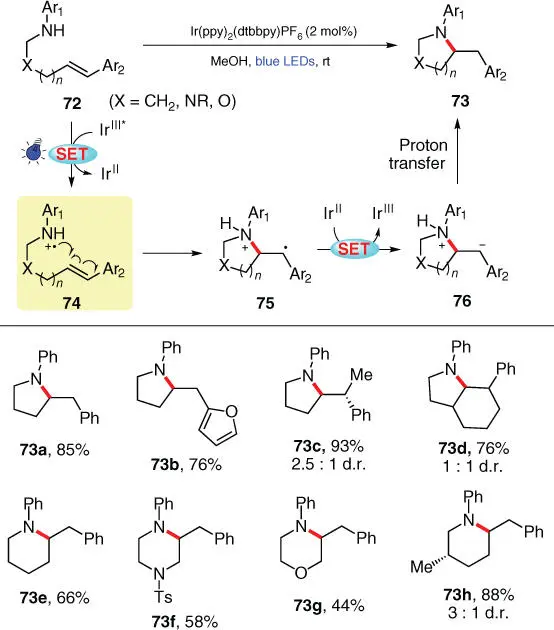
Scheme 3.13 Photocatalytic intramolecular hydroamination of olefins with aminium radical cations.
Source: Modified from Musacchio et al. [25].
With their further exploration into photocatalytic amination of alkenes, Knowles and coworkers next developed an intermolecular anti‐Markovnikov hydroamination of unactivated alkenes 78with secondary aliphatic amines 77, which is not accessible using their previous protocol ( Scheme 3.14) [26]. Various structurally diverse alkenes, including terminal and di/tri/tetra‐substituted internal ones, smoothly undergo this redox‐neutral hydroamination with a wide range of secondary alkyl amines under facile conditions ( 79a–79i). Specifically, the aminium radical cation intermediate would preferentially add to the more electron‐rich olefin when two electronically differentiated C=C bonds exist in one substrate ( 79d, 79e). Notably, this direct hydroamination method can also be applied to the intramolecular C—N bond construction ( 79j). Nevertheless, aromatic amines, α‐amino acids, and tetramethylpiperidine have been proven as inert amine partners in this reaction.
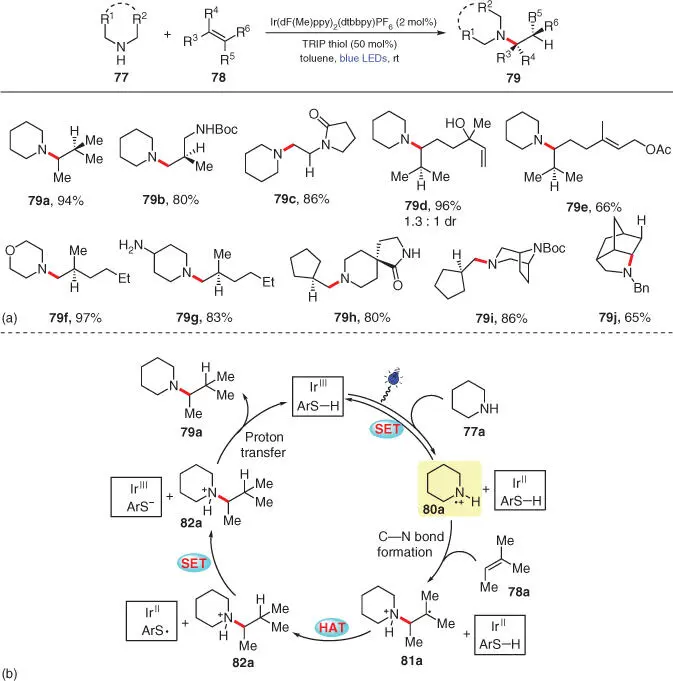
Scheme 3.14 Photocatalytic intermolecular hydroamination of unactivated olefins with secondary alkyl amines.
Source: Modified from Musacchio et al. [26].
The mechanism proposed by the authors is depicted in Scheme 3.14b. As supported by Stern–Volmer experiments, piperidine 77acan effectively quench the photoexcited Ir III* to produce the N‐centered radical cation species 80a, which then adds onto an alkene acceptor, e.g. 78a, to form a new C—N bond along with an adjacent C‐radical ( 81a). Differently from their previous amination protocol ( Scheme 3.13), intermediate 81athen abstracts a hydrogen atom from the TRIP thiol cocatalyst to deliver a closed‐shell ammonium ion 82aand a transient thiyl radical (ArS ·). Subsequently, a SET process takes place between ArS ·and the reduced Ir IIspecies to provide ArS −and the regenerated ground‐state photocatalyst Ir III. The final hydroamination product 79ais furnished after the deprotonation of 82aby anion ArS −. According to the obtained redox potentials, the final products 79could possibly be further oxidized by the excited Ir III*, but high yields of 79have still been obtained in these transformations without meaningful amounts of decomposition. The authors speculate that this outcome may result from the protective action of the thiol cocatalyst by reducing any α‐amino radical that could be engaged in further deleterious side reactions.
Again in 2019, an intermolecular anti‐Markovnikov hydroamination of unactivated olefins 84was achieved by Knowles' group, using simple primary alkyl amines 83under mild photoredox conditions ( Scheme 3.15) [27]. An identical mechanism to the former one via an aminium radical cation intermediate 86is proposed for this transformation employing a different iridium photocatalyst with divergent reactivity. Despite the presence of excess olefin, high selectivities, generally over 20 : 1, are observed for secondary over tertiary amine products.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.