Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
During the past few decades, cross‐dehydrogenative coupling (CDC) has emerged as an attractive technique, in which preinstallation of a reactive group in either of the coupling precursors is avoided, thus featuring higher atom and step economy in synthetic chemistry. Accordingly, CDC amination, which furnishes C—N bonds directly from unfunctionalized C—H bonds and N—H bonds, is acknowledged as one of the most straightforward approaches for C—N bond construction. In this field, transition‐metal‐mediated CDC aminations have been more extensively investigated, as summarized in the reviews by Patureau and coworker [2] and Chang and coworker [3]. Generally, these protocols depend on the interaction of the metal catalysts with the preinstalled directing groups on the substrates to facilitate the following amination. However, both the installation and the removal of such groups are tedious work that makes these strategies practically compromised. While, in other C–H aminations without chelation, the substrate scopes of carbon moieties are mostly limited to certain acidic C—H bonds that are more prone to activation. Nevertheless, these limitations have been overcome, wherein the direct conversion of unfunctionalized N—H and C—H bonds into C—N bonds have been achieved via reactive metal‐nitrenoid species [4]. Apart from metal chemistry, metal‐free direct oxidative C–H aminations have also been realized by the groups of Antonchick, Deboef, Chang, etc., via the mediation of hypervalent iodine reagents [5].

Scheme 3.1 C—N bond formation applying N—H bonds via photo/electrochemical methods.
Radical chemistry is reviving in organic synthesis owing to its inherently advantageous properties and has provided various alternative approaches for C—N bond formation. As is documented in one review on nitrogen‐centered radical amination, apart from some metal‐coordinated aminations, considerable progress has been advanced on photocatalytic strategies in the past years [6]. Moreover, remarkable achievements in direct electrochemical amination have also been witnessed more recently. On the basis of the active radical intermediates involved, pathways of oxidative radical aminations can be classified into three major patterns: N‐radical addition ( Scheme 3.1, path A), N‐atom nucleophilic addition ( Scheme 3.1, path B), and radical cross‐coupling ( Scheme 3.1, path C), which have enriched the methodology of C—N bond formation and broadened the substrate scope to some extent.
Accordingly, this chapter will highlight the recent advances in the photo/electrochemically mediated, radical‐involved cross‐coupling reactions of unfunctionalized N—H bonds with diverse C—H bonds, among which several cyclization or hydroamination examples with certain unsaturated carbon‐centered moieties are also included. To stimulate the design and development of more versatile and practical amination protocols, extra emphasis is put on the reaction mechanisms. The key radical intermediates involved in these transformations are highlighted in yellow boxes.
3.2 C—N Bond Formation via N‐radical Species Addition
The most familiar pathway for C—N bond formation is via the addition of in situ generated N‐radical species to unsaturated carbon‐centered moieties. Visible light photoredox catalysis has provided a large array of methodologies for C—N bond construction, and in major cases, the active N‐radical species are produced by the reductive cleavage of N—X (X = N, O, S, Cl, and Br) bonds via single‐electron transfer (SET) processes, which have already been comprehensively summarized by Xiong and Zhang [6], Xiao and coworkers [7], and Kärkäs [8]. However, unlike the reductive cleavage of weak N—X bonds, direct oxidative scission of strong N—H bonds to form N‐radical species is far more challenging because of their higher bond dissociation energies, which usually requires harsh conditions or strong stoichiometric oxidants.
3.2.1 Radical Addition to C—C Double/Triple Bonds
3.2.1.1 Amidyl Radical Addition
In the successful examples directly employing N—H bonds as the radical precursors, the N—H bonds of most aminating reagents possess relatively low p K avalues to facilitate their effective oxidation. Besides, electron‐deficient functional groups such as acyl groups and sulfonyl groups (–Ts) adjacent to the N‐atom can also significantly stabilize the generated N‐radical species. Thus, amidyl or sulfonamidyl radicals often appear as the key intermediates in the N‐radical‐based C—N bond formation.
During the past few years, proton‐coupled electron transfer (PCET) strategy has been adopted in synthetic chemistry by Knowles' group, particularly for the homolytic activation of strong N—H bonds to form N‐radical species that could further engage in various C—N bonds constructing transformations. Different from the straightforward hydrogen atom transfer (HAT) process, in a typical PCET‐enabled amination, the most crucial step is the formation of a discrete hydrogen bond complex between the N—H bond of the substrate and the catalytic Brønsted base to modulate the redox potential, which enables the following concerted exchange step of an electron and a proton to access the N‐radical species.
In 2015, employing alkene 1with a remote amidyl substituent as the substrate, Knowles and coworkers sequentially developed PCET‐based intramolecular carbo‐ and hydroamination using a combination of an iridium complex photocatalyst and a catalytic Brønsted base ( Scheme 3.2) [9]. As is proposed by the authors, the N—H bond of 1first interacts with the phosphate base, forming a hydrogen bond to lower the requirement of redox potential for the following electron transfer process. The concerted process of a one‐electron oxidation by photoexcited Ir III* species and a proton abstraction by the base provides the key amidyl radical 2, which then cyclizes onto the attached C=C bonds to form a new C—N bond with an adjacent C‐centered radical ( 3). Subsequently, the nucleophilic radical 3can further add onto an electrophilic olefin acceptor to deliver the carboamination product 4after a tandem electron/proton transfer process. On the other hand, in the presence of catalytic thiophenol, radical 3can also undergo a HAT process with the hydrogen donor to furnish the hydroamination product 5, while the resulting PhS ·is then engaged in a SET process with the Ir IIspecies to afford PhS −and regenerate ground‐state photocatalyst Ir III. The following protonation of PhS −by the conjugate acid of the phosphate base indicates the closure of both catalytic cycles.
Later in 2016, Knowles' group employed the same strategy in the selective alkylation of remote C(sp 3)—H bonds, which engages a 1,5‐HAT process with the aid of the amidyl radicals produced via PCET [10]. Simultaneously, a similar work was reported by Rovis' group [11]. Therefore, PCET activation has been proven as a powerful reaction mode for direct N‐radical formation from strong N—H bonds. Furthermore, it is also a promising platform to broadly explore this strategy in the construction of diversified C—N bonds.
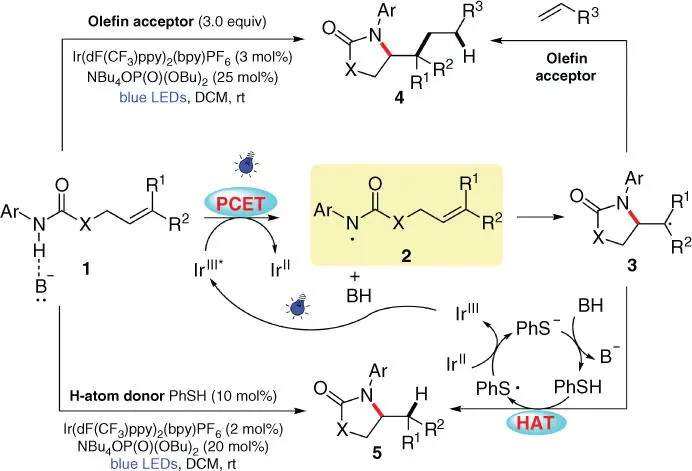
Scheme 3.2 PCET‐mediated intramolecular carbo/hyrdroamination of alkenes.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.