Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Source: Modified from Gentry and Knowles [9].
More recently, Knowles and coworkers reported a sulfonamidyl‐based hydroamination strategy, again via PCET activation ( Scheme 3.3) [12]. Hydroamination products 7and 10could be smoothly furnished from intramolecular cyclization of 6and intermolecular reaction between 8and 9, respectively, employing 2,4,6‐triisopropyl‐thiophenol (TRIP thiol) as the HAT catalyst via the same pathway as depicted in Scheme 3.2. Apart from the broad substrate scope demonstrated, a series of tandem amination/C–H alkylation sequences were performed to highlight the synthetic versatility ( Scheme 3.3b). The terminal alkene 11was first subjected to the intermolecular anti‐Markovnikov hydroamination with p ‐methoxyphenyl (PMP) sulfonamide 12to afford the alkylated sulfonamide 13, and the newly installed secondary sulfonamide was then activated in a second oxidative PCET event using the same Ir/phosphate pair, leading to the site‐selective abstraction of the δ‐C—H bond to afford a carbon‐centered radical that can be further trapped by an electron‐deficient olefin 14to afford the final product 15.
The reviving synthetic organic electrochemistry has been providing environmentally benign alternatives to the traditional synthetic methods because of its generally high atom economy and good functional group compatibility. In the electrochemical C—N bond formation from unfunctionalized N–H/C–H precursors, a series of advances have been achieved by Xu's group in the recent years. In 2016, Xu's group disclosed an electrocatalytic hydroamination of alkenes, in which an inexpensive organometallic reagent ferrocene was employed as a redox catalyst to enable the direct generation of amidyl radicals from N ‐aryl amides 16( Scheme 3.4) [13]. Based on the optimization of the reaction conditions, a mixed solvent tetrahydrofuran (THF)/MeOH in 5 : 1 ratio is demonstrated as the optimal choice, while no reaction takes place in the single solvent MeOH, which is also supported by the cyclic voltammetry (CV) observations.
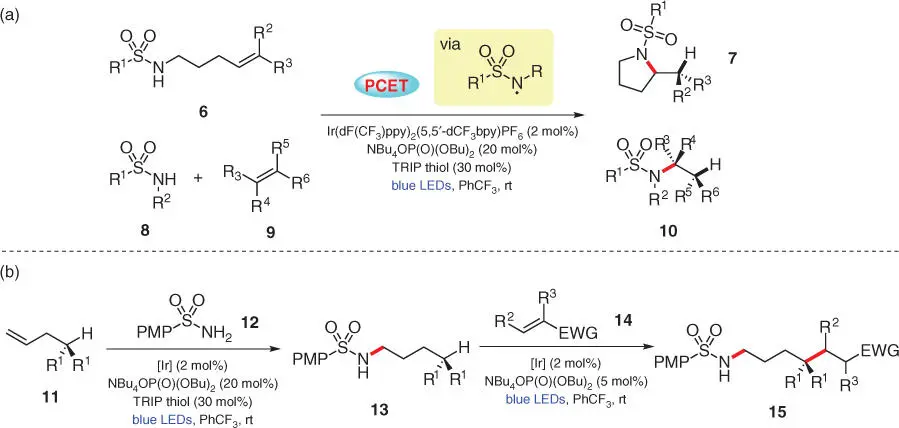
Scheme 3.3 PCET‐mediated intra/intermolecular amination of alkenes. (a) Hydroamination of alkenes with primary or secondary sulfonamides. (b) Tandem amination/C–H alkylation.
Source: Modified from Zhu et al. [12].
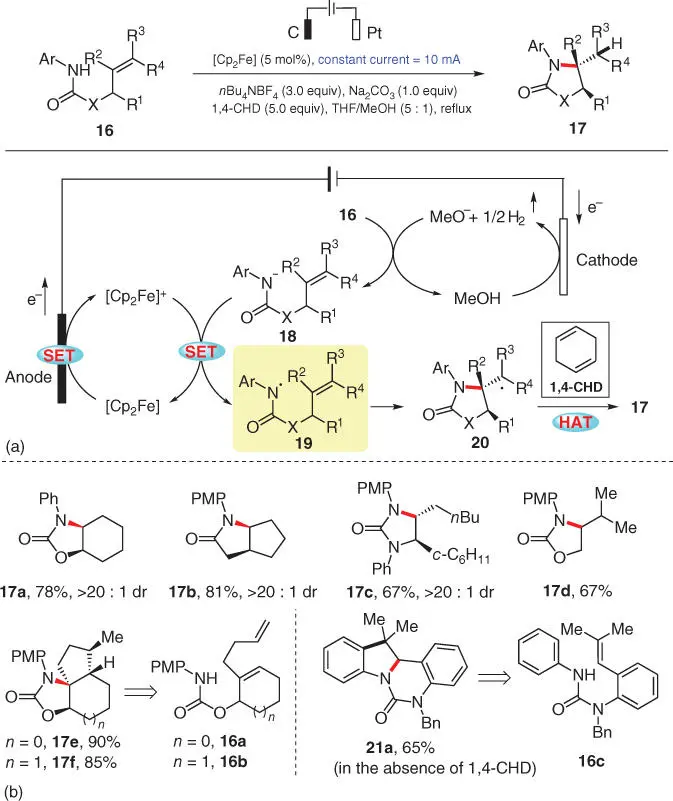
Scheme 3.4 Electrocatalytic intramolecular hydroamination of alkenes.
Source: Modified from Zhu et al. [13].
As depicted in their mechanistic proposal ( Scheme 3.4a), this reaction is supposed to begin with the anodic oxidation of ferrocene ([Cp 2Fe]) and the simultaneous cathodic reduction of cosolvent methanol. Subsequently, the electrochemically generated base MeO −deprotonates the amide group of substrate 16to afford anionic intermediate 18, which can be easily oxidized by [Cp 2Fe] +to provide the key amidyl radical 19, along with the regenerated mediator [Cp 2Fe]. Radical 19then cyclizes onto its tethered alkene to furnish intermediate 20, which further acquires a hydrogen atom from the H‐atom donor 1,4‐cyclohexadiene (1,4‐CHD) to yield the desired product 17. The scope exploration reveals that various carbamates, ureas, and amides can serve as viable substrates, providing the corresponding products in good to high yields under the standard conditions ( Scheme 3.4b, 17a–17d). Notably, in the reactions of diene substrates 16aand 16b, tandem cyclization processes occurred to furnish polycyclic products 17eand 17fwith high efficiency. Moreover, under standard conditions but in the absence of 1,4‐CHD, substrate 16cfinally turns into indoline 21aafter an oxidative termination step.
Again, Xu and coworkers employed the same electrochemical N‐radical formation strategy in the synthesis of highly functionalized (aza)indoles 23through intramolecular N‐radical species addition of 22to their remotely attached alkynyl moieties ( Scheme 3.5) [14]. In this transformation, ferrocene is also selected as the redox mediator upon anodic oxidation, and the simultaneous cathodic reduction converts the cosolvent methanol into MeO −base and H 2gas. Subsequently, a SET process between the oxidized [Cp 2Fe] +and anion 24from deprotonation of 22produces an electron‐deficient, N‐centered radical 25and meanwhile regenerates [Cp 2Fe]. Radical 25would then preferentially undergo a 6‐exo‐dig cyclization to vinyl radical 26, followed by a second cyclization to give the delocalized radical 27, as also supported by the density functional theory (DFT) calculations. Finally, rearomatization of 27after an oxidation/deprotonation sequence delivers the final product 23. A broad substrate scope is demonstrated under the standard conditions, exhibiting high functional group tolerance ( Scheme 3.5b). Notably, the late‐stage modification of ethinyl estradiol proceeds smoothly to deliver the indole‐functionalized estradiol 23a. The acid/base‐sensitive chiral amino esters ( 23b, 23c) as well as a free alcohol ( 23d) are also well tolerated under the electrolysis.
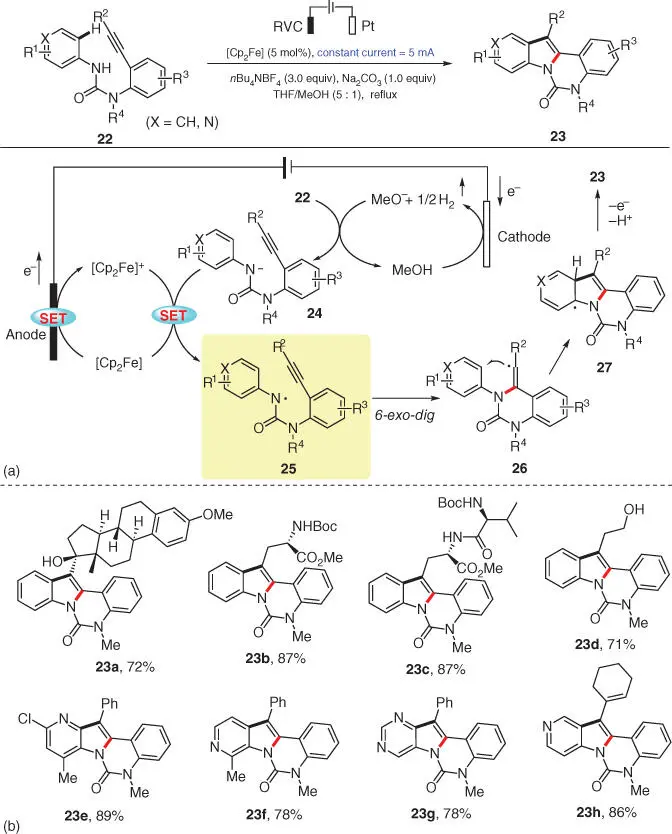
Scheme 3.5 Electrochemical synthesis of multifunctionalized (aza)indoles. RVC, reticulated vitreous carbon.
Source: Modified from Hou et al. [14].
In 2017, Xu and coworkers adopted substrate 28with a polysubstituted alkene moiety to achieve an electrochemical aza‐Wacker‐type cyclization, wherein the key amidyl radical 29is generated by direct anodic oxidation of the amidyl N—H bond without the aid of a base ( Scheme 3.6) [15]. The following intramolecular cycloaddition of radical 29to its pendant alkenyl group furnishes C‐centered radical 30, which is more prone to suffer further oxidation rather than H‐atom abstraction, to deliver the corresponding cation 31. The desired product 32is finally generated from cation 31after deprotonation. This electrochemical protocol achieves the challenging intramolecular oxidative amination of sterically demanding alkenes in the absence of a metal catalyst and is proven compatible with a wide range of carbamates ( 32a–32d), amides ( 32e, 32f), and ureas ( 32g, 32h). Moreover, product 32iwith a steroid‐based core is smoothly afforded in 70% yield from a tetrasubstituted alkene under electrolysis, demonstrating the extra synthetic potential of this protocol.
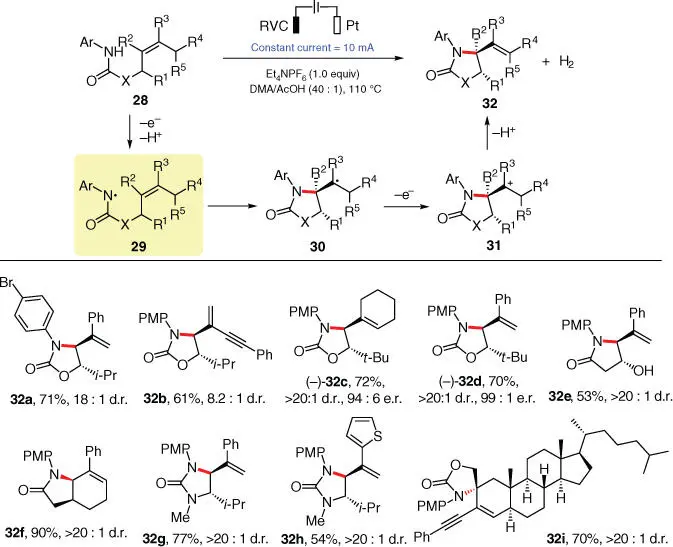
Scheme 3.6 Electrochemical intramolecular oxidative amination of tri‐ and tetra‐substituted alkenes.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.