Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
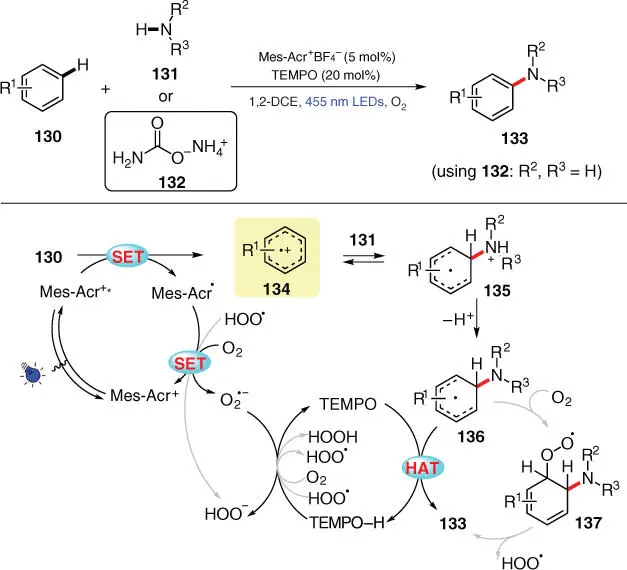
Scheme 3.24 Site‐selective aromatic C–H amination via photoredox catalysis.
Source: Modified from Romero et al. [37].
The direct oxidative C–N cross‐coupling between arenes and azoles has also been realized with other photocatalytic systems. In 2017, Lei's group employed a dual catalytic system combining an acridinium photocatalyst with a Co‐based cocatalyst for the C—N bond formation to access N ‐arylazoles ( Scheme 3.25) [38]. This sustainable protocol does not require any sacrificial oxidant, and the released H 2gas is the only by‐product. Various aromatics including substituted benzenes, biphenyls, and anisole derivatives are smoothly aminated under the standard conditions. Based on the kinetic isotope effect (KIE) experiments and other mechanistic studies, the reaction begins with the oxidation of 138by the photoexcited Mes‐Acr +*, which leads to the formation of Mes‐Acr ·and radical cation 141. The ground‐state photocatalyst Mes‐Acr +is then regenerated by single‐electron oxidation of Mes‐Acr ·by Co IIIcatalyst. On the other hand, nucleophilic attack of amine 139to radical cation 141forms radical intermediate 142after deprotonation, which subsequently undergoes a single‐electron oxidation by Co IIto form cation 143. The final product 140is delivered after deprotonation of 143.
In the same year, Pandey's group reported a regioselective method for the direct C(sp 2)–H amination of anisoles with different azoles under visible light conditions using Selectfluor as an external oxidant ( Scheme 3.26) [39]. As a strong oxidant, Selectfluor is capable of oxidizing the photoexcited Ru II* to its higher valence state Ru IIIand meanwhile furnishing radical cation 147. The resulting Ru IIIthen readily undergoes a SET reduction with the electron‐rich arene 144to complete the photocatalytic cycle and simultaneously generates radical cation 148, which is further attacked by the amine nucleophile 145to give radical intermediate 149. After deprotonation of 149by 2,6‐lutidine and H‐atom abstraction of 150by radical 147, the desired amination product 146is finally obtained.
In 2019, an electrochemical alternative was disclosed by Wu and coworkers, wherein exclusive ortho‐selectivity of amination was achieved in the most cases, under metal‐ and oxidant‐free conditions ( Scheme 3.27) [40]. During the optimization of the reaction conditions using anisole 151aand pyrazole 152a, the authors found that the addition of trifluoroacetic acid (TFA) could effectively enhance the ortho‐regioselectivity. Similarly, in the subsequent scope evaluation, it was also observed that reactions with TFA provided significantly higher o : p selectivity than the corresponding ones without it ( 153a, 153d, and 153e). The proposed mechanism is depicted in Scheme 3.27b. Anisole 151ais preferentially oxidized to provide the radical cation 154aat the anode, after which pyrazole 152aas a nucleophile attacks 154ato form the ortho‐aminated intermediate 156avia a putative intermediate 155awith TFA‐assisted hydrogen bond interaction. Final product 153ais formed after deprotonation and rearomatization of 156a.
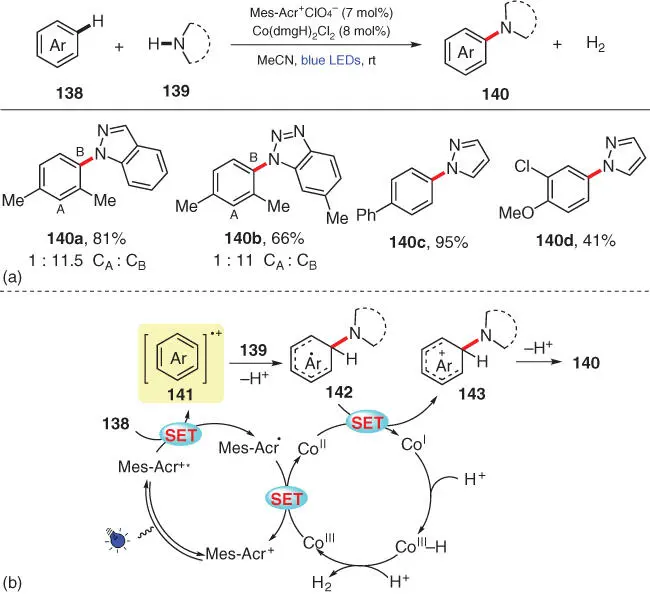
Scheme 3.25 Photoinduced oxidant‐free C–H amination of arenes with azoles.
Source: Modified from Niu et al. [38].
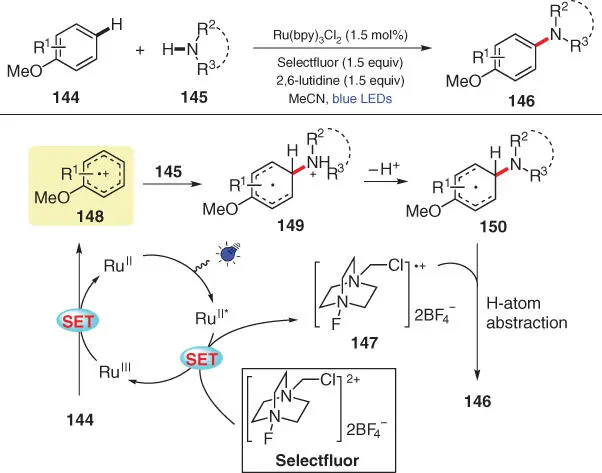
Scheme 3.26 Selective C–H amination of electron‐rich arenes via photoredox catalysis.
Source: Modified from Pandey et al. [39].
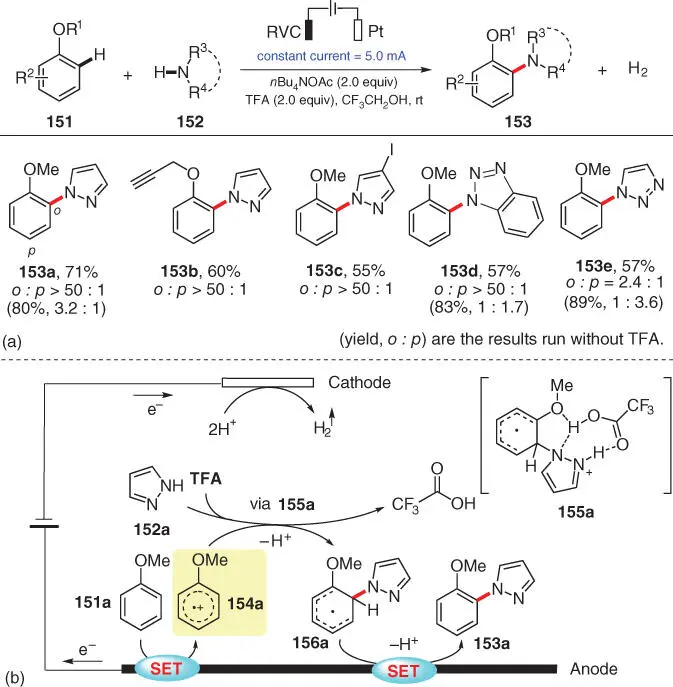
Scheme 3.27 Electrochemical ortho‐amination of aromatic C—H bonds with azoles.
Source: Modified from Wang et al. [40].
Electron‐rich pyrroles and thiophenes can easily be oxidized into their corresponding radical cations to undergo further C—N bond formation processes via N‐atom nucleophilic addition pathway. In 2016, König and coworkers realized a photocatalytic C(sp 2)–H sulfonamidation of pyrroles to reach a range of N ‐(2‐pyrrole)sulfonamides 159( Scheme 3.28) [41]. In this transformation, N‐substituted pyrrole 157is first oxidized by the photoexcited organic dye Mes‐Acr +into its corresponding radical cation 160, which is attacked by strong anionic nucleophile 161,resulting from deprotonation of sulfonamide 158to generate radical intermediate 162. The final amination product 159is afforded via a HAT process with O 2 ·−or alternatively through further oxidation and deprotonation sequence of 162.
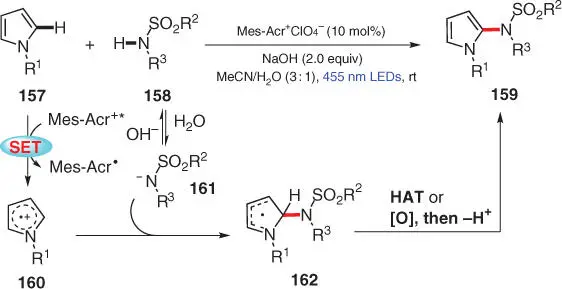
Scheme 3.28 Direct C2‐sulfonamidation of pyrroles via visible‐light photoredox catalysis.
Source: Modified from Meyer et al. [41].
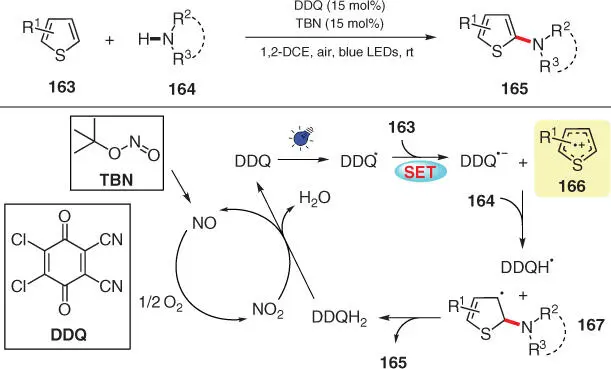
Scheme 3.29 DDQ‐mediated C2‐amination of thiophenes via visible‐light photoredox catalysis.
Source: Modified from Song et al. [42].
In the following year, Lei and coworkers disclosed a photocatalytic C(sp 2)–H amination of thiophenes with azoles, employing 2,3‐dicyano‐5,6‐dichlorobenzoquinone (DDQ) as an organic photocatalyst, tert ‐butyl nitrite (TBN) as an electron transfer mediator, and aerobic oxygen as the oxidant ( Scheme 3.29) [42]. The photoexcited catalyst DDQ* upon visible light irradiation possesses a high oxidation potential ( E red= 3.18 V vs. saturated calomel electrode [SCE]) and thus can easily oxidize thiophene 163into radical cation 166. Nucleophilic addition by azole 164and the following proton donation to DDQ ·−convert radical cation 166into radical intermediate 167, which undergoes further HAT process with the formed DDQH ·to yield the desired products 165and DDQH 2. To complete the photocatalytic cycle, DDQH 2then participates in another cycle involving TBN to regenerate the original photocatalyst DDQ.
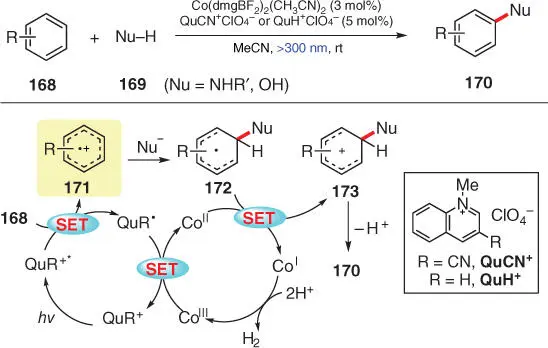
Scheme 3.30 Photocatalytic benzene C–H amination and hydroxylation with hydrogen evolution.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.