Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Source: Modified from Hu et al. [47].
3.3.3 Activated C(sp 3)—H Bond Amination
In the early years, C(sp 3)–H amination reactions were mainly achieved via transition metal catalysis or with hypervalent iodine reagents. Recently, quite a few methods for CDC amination of activated C(sp 3)—H bonds, such as benzylic C(sp 3)–H and C(sp 3)–H in α-position to a nitrogen atom, have been realized via the formation of the corresponding cationic species and subsequent addition of nitrogen nucleophiles by means of photo/electrochemistry.
3.3.3.1 Benzylic C(sp 3)—H Bond Amination
As an example for the direct amination of benzylic C(sp 3)—H bonds, Pandey's group disclosed a photocatalytic CDC amination of alkylbenzenes 196with N ‐methoxyamides 197under metal‐free and external oxidant‐free conditions ( Scheme 3.34) [48]. The 410 nm wavelength visible light source is obtained by using a combination of Pyrex and a CuSO 4:NH 3solution filter from a 450 W Hanovia medium pressure lamp. Compound 9,10‐dicyanoanthracene (DCA) is employed as a photosensitizer, which facilitates an array of C(sp 3)–H amination reactions between 196and 197with good functional group tolerance ( 198a– 198f). Notably, this protocol is also applicable to intramolecular C—N bond construction to provide five‐membered rings ( 198g). The mechanistic proposal is depicted in Scheme 3.34b. First, the single‐electron oxidation of 197by the photoexcited DCA* yields the N ‐methoxyamide radical cation 199, which then converts into radical intermediate 200after deprotonation. The following HAT process from the benzylic position of alkylbenzene 196to radical 200regenerates 197and meanwhile gives benzylic radical 201. The subsequent oxidation of 201generates benzylic cation 202, which further reacts with 197to deliver the desired amination product 198. The results of control experiments and some experimental observations add credit to this putative mechanism. For example, the existence of N‐centered radical intermediate 200is suggested by the formation of its dimeric product, while benzylic radical 201is captured by TEMPO in a radical trapping experiment. To demonstrate the intermediacy of benzylic cation 202, the authors performed the model reaction using a mixture of MeCN/AcOH (9 : 1) as the solvent, which provides the desired 198in diminished yield, along with the by‐product benzyl acetate in 10% yield.
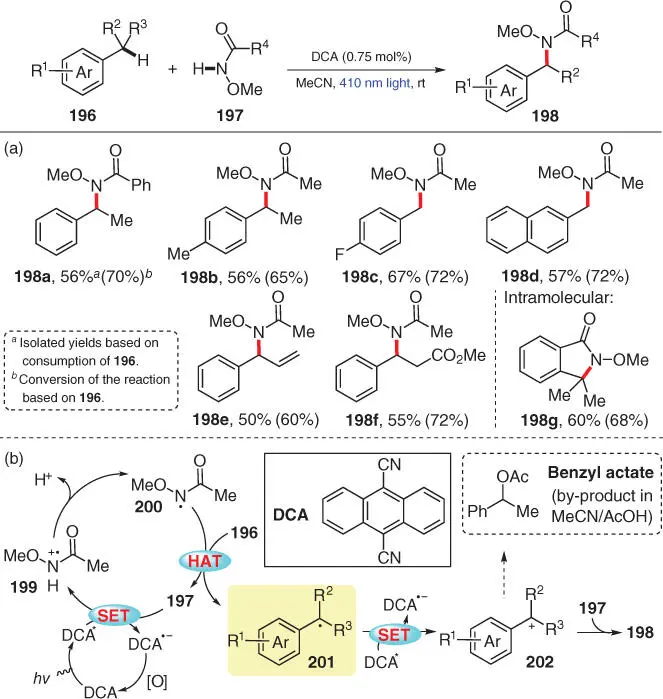
Scheme 3.34 Visible‐light‐enabled direct benzylic C(sp 3)–H amination.
Source: Modified from Pandey and Laha [48].
Alkyl‐substituted arenes with available benzylic C(sp 3)—H bonds tend to undergo direct C–H functionalization at the benzylic position via the preliminarily formed arene radical cation intermediate upon single‐electron oxidation. In 2016, Pandey et al. developed a visible‐light‐mediated cross‐dehydrogenative amination of benzylic C(sp 3)—H bonds with azole derivatives, employing an Ir‐based photocatalyst and bromotrichloromethane (BrCCl 3) as an oxidative quencher ( Scheme 3.35) [49]. Upon photoexcitation, the excited catalyst (Ir III*) first undergoes a single‐electron oxidation by BrCCl 3, affording the high‐valence catalyst Ir IVand a trichloromethyl radical ·CCl 3. Subsequently, arene substrate 203is oxidized by the Ir IVspecies to generate radical cation 206, along with the regenerated ground‐state Ir IIIphotocatalyst. Then, radical ·CCl 3as a competent H‐atom abstractor engages in a HAT process with intermediate 206to provide benzylic cation 207, which further reacts with nucleophile 204to deliver the final product 205. Moreover, it is possible to trap cation 207with moisture to in situ generate the benzylic alcohol intermediate, which is prone to further oxidation to furnish the corresponding carbonyl compound.
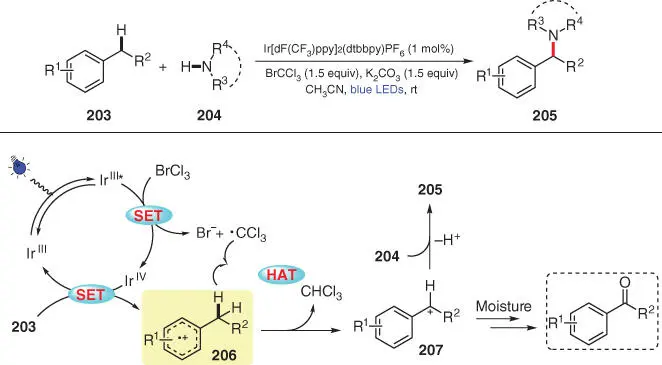
Scheme 3.35 Benzylic C–H amination via visible‐light photoredox catalysis.
Source: Modified from Pandey et al. [49].
3.3.3.2 N‐α‐C(sp 3)—H Bond Amination
The activated C(sp 3)–H adjacent to a nitrogen atom is also prone to photo/electrochemical oxidation and to subsequent nucleophilic amination. In 2019, Zeng and coworkers reported an electrochemical dehydrogenative imidation of N ‐methyl benzylamines 208with phthalimides 209to obtain various phthalimide‐protected gem ‐diamines 210( Scheme 3.36) [50]. Notably, the amination occurs regioselectively at the methyl group rather than at the benzylic position of each N ‐methyl benzylamine. Apart from multiple phthalimides with different substituents ( 210a– 210e), certain triazoles are also viable amine sources ( 210f). Based on CV studies, revealing that benzylamine 208ais easier to be oxidized than phthalimide 209a, a plausible mechanism is proposed as shown in Scheme 3.36b. The reaction is initiated by anodic oxidation of 208ato give the radical cation 211a, which undergoes deprotonation and further oxidation to generate the iminium intermediate 212a. Meanwhile, MeOH is reduced at the cathode to give MeO −and H 2. Simultaneously, 209ais deprotonated by MeO −to give the anionic intermediate 213a, which then undergoes a Mannich‐type addition to 212agenerating product 210a.
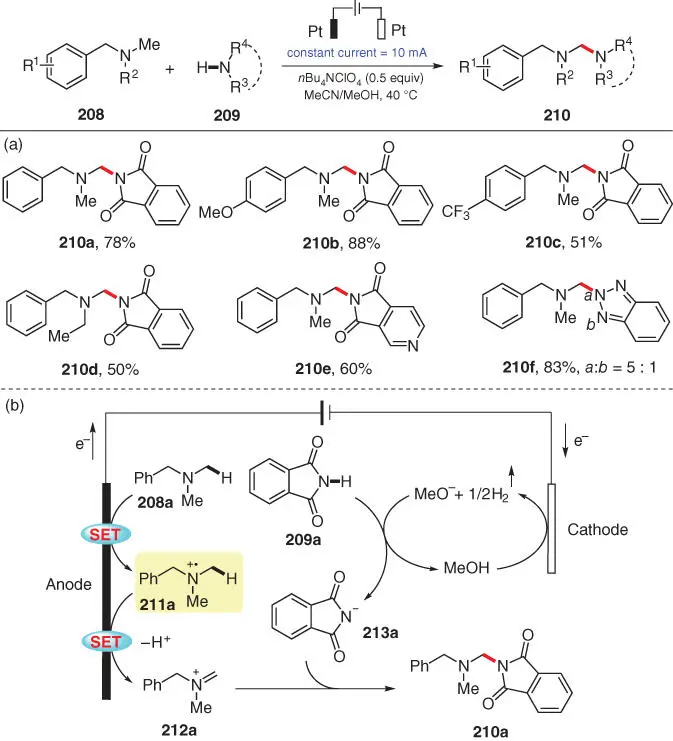
Scheme 3.36 Electrochemical dehydrogenative imidation of N ‐methyl‐substituted benzylamines.
Source: Modified from Lian et al. [50].
Almost simultaneously, Lei's group also reported an electrochemical direct imidation of N ‐methyl anilines with hydrogen evolution under acidic conditions ( Scheme 3.37) [51]. Acetic acid plays a key role in this reaction, other acids such as HCOOH, H 2SO 4, and TsOH could not enable the reaction to proceed, while n PrCOOH led to reduced efficiency. Cyclic amides, heteroatom‐containing amides, and succinimides are well tolerated in this transformation and afford products in moderate to good yields ( 216a–216e). The proposed reaction mechanism is nearly identical to the previous one.
Recently, an elegant visible‐light‐driven intramolecular C–N cross‐coupling reaction was disclosed by Zheng's research group under mild and metal‐free conditions ( Scheme 3.38) [52]. Various polycyclic quinazolinone derivatives ( 221a–221fand 223a– 221d), including natural products tryptanthrin, rutaecarpine, and their analogs ( 223b–221d), could be smoothly furnished via this protocol. Further demonstration of gram‐scale synthesis and solar‐driven transformation proves the potential of this strategy for practical applications.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.