Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:

Scheme 3.37 Electrochemical dehydrogenative imidation of N ‐methyl substituted anilines.
Source: Modified from Wang et al. [51].
The mechanistic proposal, on the basis of control experiments, DFT calculations, UV–vis spectroscopy, and electron paramagnetic resonance (EPR) studies, as well as X‐ray characterization of the key intermediate, is outlined in Scheme 3.39with 221ataken as the example. The reaction is initiated by a photoisomerization of 220aupon irradiation with visible light to produce the photoisomer 222a, which displays absorbance around 395 nm and has a short lifetime. The long‐lived photoisomer complex 223ais then generated by the coordination of ( R )-(-)-1,1'-binaphthyl-2,2'-diyl hydrogenphosphate [( R )‐BPA] through hydrogen bonding, which can induce the SET process under irradiation with visible light to generate the radical pair 224a. The resulting superoxide radical anion abstracts a hydrogen atom from 224ato produce amino radical 225a, which undergoes intramolecular 1,6‐HAT process to generate radical 226a. Then, the radical recombination of 226aand HOO ·forms the key intermediate hydroperoxide 227a, which is easily converted into iminium ion 228aunder acidic conditions provided by the Brønsted acid ( R )‐BPA. Finally, the polycyclic quinazolinone product 221ais formed through intramolecular nucleophilic addition with release of H 2O 2. As an alternative pathway, the oxidation by‐product 229a, formed from the hydroperoxide 227a, can be efficiently converted into 221aunder the standard reaction conditions, as proven by a control experiment.
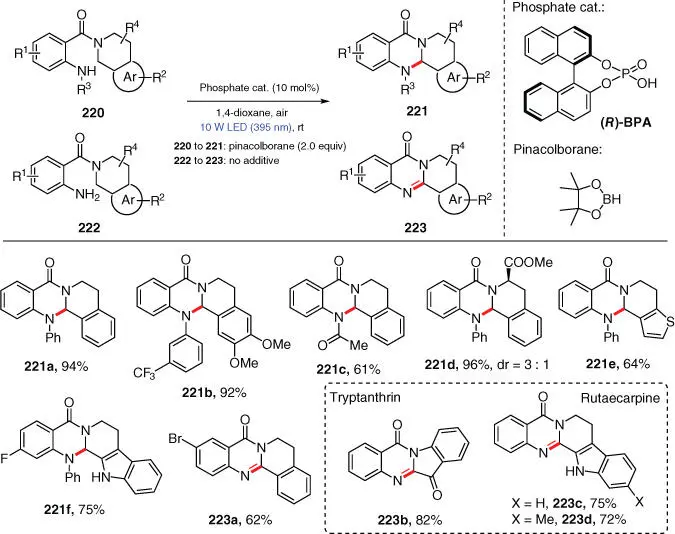
Scheme 3.38 Light‐driven direct intramolecular C–N cross‐coupling.
Source: Modified from Jing et al. [52].
3.4 Amination via Radical Cross‐coupling
The last pattern for the radical‐involved C—N bond formation is the cross‐coupling of C‐ and N‐radicals, which is relatively undeveloped compared with the aforementioned two pathways, presumably because of the challenge to provide an appropriate environment for the simultaneous generation of both C‐ and N‐radicals. Moreover, the generated C‐ and N‐radical in the same system are supposed to obey to the principle of persistent radical effect (PRE) to furnish the desired C–N cross‐coupling product rather than the homocoupling ones.
3.4.1 Aryl C(sp 2)—N Bond Formation via Radical Cross‐coupling
Among the reported C—N bond construction methods directly from aromatic C(sp 2)—H bonds and N—H bonds via radical cross‐coupling pathway, both of the coupling partners are required to be redox active to produce their corresponding radical intermediates upon oxidation. Therefore, in most cases, electron‐rich arenes such as phenols and anilines are chosen as the aryl radical precursors. As for the amine partner, electron‐rich diarylamines and azoles are commonly used. Sulfonamides after deprotonation can also turn into the corresponding radicals upon oxidation, as previously described. For these amine sources, the stabilizing effect for the N‐radicals also appears to be crucial for the success of the radical cross‐coupling.
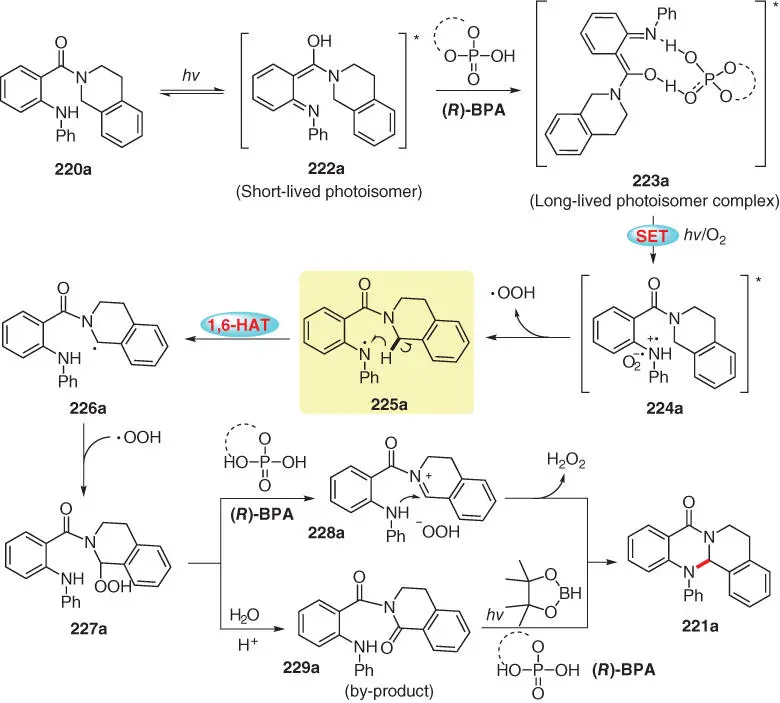
Scheme 3.39 Mechanistic proposal for the light‐driven direct intramolecular C–N cross‐coupling.
3.4.1.1 Aryl C(sp 2)—N Bond Formation Using Diarylamines
In 2016, Xia and coworkers developed a cross‐dehydrogenative C–N coupling between phenols 230including trimethylsilyl (TMS)‐protected ones and phenothiazines 231to afford a variety of ortho‐aminated phenols 232employing K 2S 2O 8as the external oxidant under mild visible light photoredox conditions ( Scheme 3.40) [53]. This protocol requires no additional photocatalyst, which is probably because of the inherent properties of phenothiazines ( 231) that can absorb visible light to enable the following energy transfer process. Notably, instead of inhibiting the amination or lower the reaction efficiency, additional radical scavenger TEMPO dramatically improves the yield of desired product 232a. Moreover, the reaction system of 3,4‐dimethoxyphenol ( 230b) and 2‐methoxylphenothiazine ( 231b), which only yields 232cas a homocoupling product of 231bunder the original conditions, does furnish the desired C–N cross‐coupling product 232bwith extra 2.0 equiv of TEMPO added. On the basis of some relevant literature, a hypothesis that TEMPO can covalently and reversibly prolong the lifetime of the transient N‐radicals is reasonably proposed by the authors. Mechanistically, under the irradiation of visible light, oxidant K 2S 2O 8is capable of oxidizing both phenol and phenothiazine to provide C‐radical 235and N‐radical 236, respectively. The following radical–radical cross‐coupling process generates intermediate 237, which further tautomerizes into the final amination product 232.
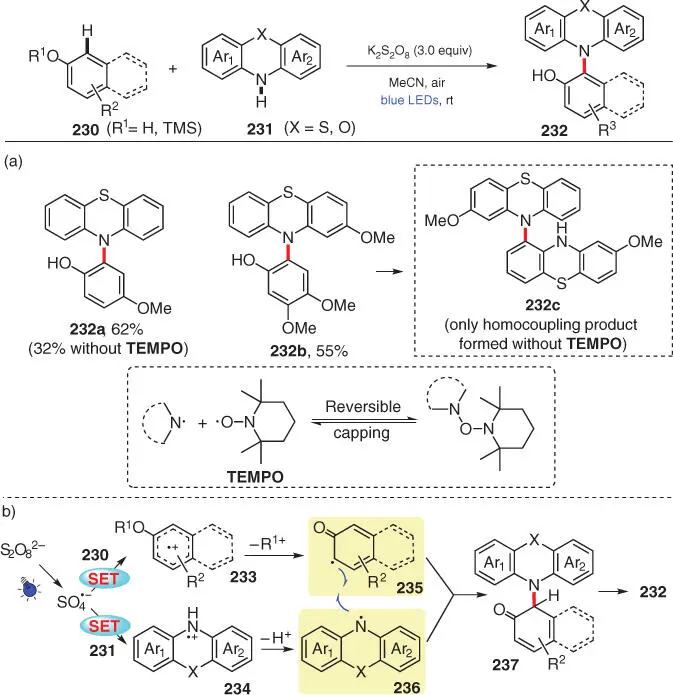
Scheme 3.40 Visible‐light‐promoted CDC amination of phenols and phenothiazines. (a) Reactions with additional 2.0 equiv of TEMPO. (b) The mechanistic proposal.
Source: Modified from Zhao et al. [53].
However, the amine scope of this catalyst‐free amination is limited to S‐ or O‐containing cyclic diarylamines, which does not include acyclic ones because they are incapable of absorbing visible light and possess higher oxidation potentials. To accommodate these substrates, Xia's group subsequently developed another photocatalytic strategy to enable the CDC amination of phenols 238with acyclic diarylamines 239employing the organic salt 2,4,6‐triphenylpyrylium (TPT) as a photocatalyst ( Scheme 3.41) [54]. This protocol is proven compatible with a wide range of electron‐rich acyclic diarylamines and phenols including the natural product (+)‐δ‐tocopherol ( 240a–240c). Notably, phenothiazine or phenoxazine substrates, which require prolonged time to complete the reaction using the previous method, convert much faster under these conditions. As demonstrated by a series of mechanistic studies including EPR spectroscopy and fluorescence quenching experiments, diaryl amine 239is able to reductively quench the photoexcited catalyst TPT*, generating the corresponding radical cation 241along with reduced TPT ·−. A radical chain propagation process is indicated by the determined quantum yield (Φ = 19), which is proposed to take place between phenol 238and N‐centered radical cation 241to provide phenol radical cation 242regenerated amine 239. An alternative pathway to reach 242is through the SET oxidation of 238by persulfate. Finally, radicals 244and 243formed from deprotonation couple with each other to deliver the cross‐coupling product 240.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.