Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Source: Modified from Zheng et al. [43].
Apart from the most widely used N–H nucleophile azoles, ammonia as an abundantly available and cheap reagent has also attracted attention of researchers, and has been exploited in nucleophilic CDC amination. In 2016, Wu and Tung and coworkers developed a dual catalytic cross‐coupling between aromatic C—H bonds and ammonia or water under visible‐light conditions to furnish anilines or phenols with the evolution of hydrogen gas ( Scheme 3.30) [43]. N ‐Methylquinolinum salts QuCN +ClO 4 −and QuH +ClO 4 −are employed as the photocatalysts (QuR +), whose the excited states possess enough oxidizing power ( E red* = 2.72 and 2.46 V vs. SCE, respectively) to accept an electron from benzene ( E ox= 2.48 V vs. SCE) to furnish the reduced photocatalyst QuR ·along with the crucial radical cation 171. Subsequently, the former is oxidized by Co IIIcatalyst to regenerate the ground‐state photocatalyst QuR +; meanwhile, the latter is captured by a nucleophile to form radical 172, which further donates an electron to Co IIto produce cationic species 173. The final product 170is formed after further deprotonation of 173. On the basis of CV studies, the redox potential values demonstrate that product 170is also capable of quenching the excited photocatalyst QuR +*, which may result in its further functionalization. However, mono‐functionalized benzene is obtained as the only product, and no multiaminated or hydroxylated products can be detected even after prolonged reaction time. A hypothesis for this result, advanced by the authors, is that the rapid back‐electron transfer from the reduced QuR ·to the formed radical cation of 170protects product 170from overoxidation.
3.3.2 Olefinic C(sp 2)—H Bond Amination
As good radical acceptors, alkenes are capable of trapping various radicals to deliver the corresponding addition products. Nonetheless, the direct nucleophilic amination of alkenes depends on the prior oxidation of the C=C bonds to form the crucial C‐centered radical cation intermediates, which is followed by subsequent nucleophilic addition of N–H partners.
During the past decade, Nicewicz's group has developed a variety of visible‐light‐induced anti‐Markovnikov hydrofunctionalization protocols for alkenes via the addition of nucleophiles to the in situ generated C‐centered radical cations, employing the strongly oxidizing acridinium salts as photocatalysts. Based on their previous work on the intramolecular hydroamination of alkenes 174using a combination of catalytic Mes‐Acr +BF 4 −and thiophenol as the photocatalyst and H‐atom donor, respectively ( Scheme 3.31a) [44], they later reported an intermolecular anti‐Markovnikov hydroamination of alkenes by replacing the thiophenol cocatalyst with a combination of phenyl disulfide and 2,6‐lutidine ( Scheme 3.31b) [45]. Therein, trifluoromethanesulfonamide (TfNH 2) and azoles ( 177) are employed as the N–H nucleophiles to approach a variety of phenethylamine derivatives 178. In line with their previous work on nucleophilic alkene functionalization, the authors suggest that the key step is the SET process between alkene 176and the photoexcited acridinium (Mes‐Acr +*), which results in the generation of key radical cation intermediate 179. Subsequently, nucleophiles TfNH 2or 177undergo reversible addition to the least‐substituted side of 179to form the stabilized C‐radical 180, which may further undergo a HAT process with a hydrogen atom donor, presumably thiophenol, to deliver the desired product 178. On the other hand, the putative HAT reagent thiophenol is supposed to come from phenyl disulfide after a homolysis/single‐electron reduction/protonation sequence.
In 2017, Lei and coworkers developed an external oxidant‐free photocatalytic CDC amination and etherification of alkenes with azoles and alcohols using a dual catalytic system combining photocatalyst Mes‐Acr +ClO 4 −and a Co‐based cocatalyst ( Scheme 3.32) [46]. The single‐electron oxidation of alkene 181by the photoexcited Mes‐Acr +* is considered as the key initial step, providing Mes‐Acr ·and radical cation species 186. Subsequently, the employed nucleophile 182(or 183) reacts with 186to form the more stable radical intermediate 187, which is further oxidized into the corresponding cationic species 188by either Mes‐Acr +* or Co III. Then, the final anti‐Markovnikov product 184(or 185) is delivered after deprotonation of 188. As for the Co catalytic cycle, the reduced Co IIundergoes further reduction by Mes‐Acr ·to yield Co Ispecies, which is protonated twice and then releases one molecule H 2to regenerate the Co IIIspecies.
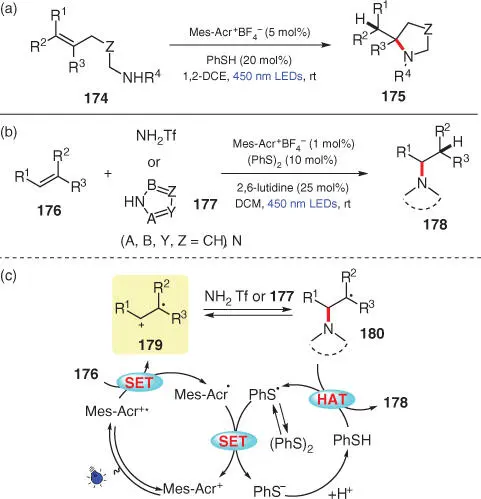
Scheme 3.31 Anti‐Markovnikov hydroaminations of alkenes by two component organic photoredox systems.
Source: Modified from Nguyen and Nicewicz [44].
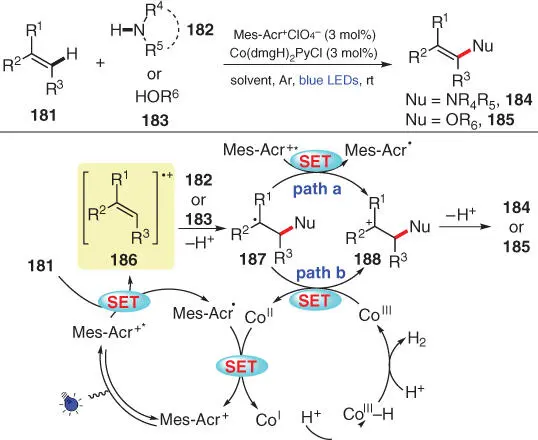
Scheme 3.32 Photocatalytic dehydrogenative cross‐coupling of alkenes with azoles and alcohols via a dual catalytic system.
Source: Modiifed from Yi et al. [46].
In 2018, again in Lei's laboratory, a photoinduced oxidative [4+2] annulation of imines and alkenes was developed by utilizing a dual photoredox/cobaloxime catalytic system in a mixed solvent of 1,2‐DCE and hexafluoroisopropanol (HFIP) ( Scheme 3.33) [47]. This method not only obviates the application of stoichiometric oxidants but also exhibits excellent atom economy by generating hydrogen gas as the only by‐product, achieving high regioselectivity and trans ‐diastereoselectivity even if alkenes are employed in Z / E mixtures. On the basis of Stern–Volmer studies and CV experiments, a plausible mechanism is proposed. Firstly, single‐electron oxidation of 190by the excited‐state photosensitizer leads to the generation of the alkene radical cation intermediate 192and to the reduced Mes‐Acr ·. Subsequently, the nucleophilic attack of 189furnishes the radical 193after deprotonation, in which the C=C single bond prefers a more stable trans‐configuration upon rotation as proposed by the authors. Next, the radical cyclization of 193gives the intermediate 194, which is further oxidized to furnish 195. After elimination of a proton, the desired 3,4‐dihydroisoquinoline product 191is afforded. On the other hand, the reduced photosensitizer is oxidized by the Co IIIspecies to complete the photoredox catalytic cycle. As for the cobalt side, the Co IIspecies is reduced to form a Co Iintermediate, which can be protonated to produce Co III‐hydride. The Co III‐hydride species would then release H 2upon the interaction with protons.
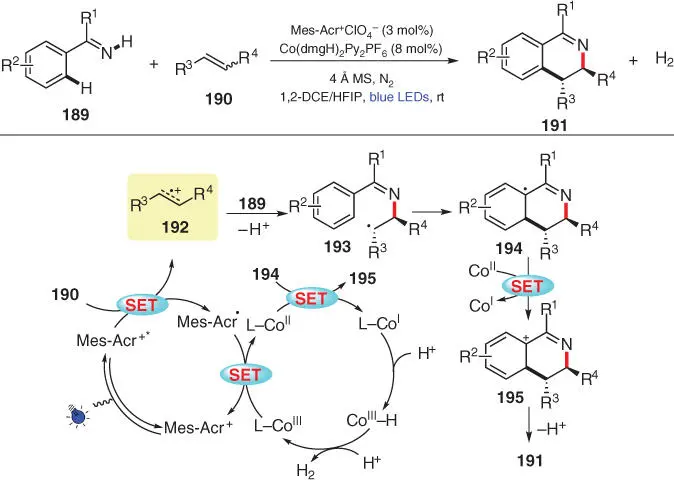
Scheme 3.33 Photoinduced oxidative [4+2] imine/alkene annulation with H 2liberation.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.