Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
The ability of amidyl radicals to undergo 1,5‐HAT has also been exploited as part of a strategy, leading to remote sp 3C–H arylated products 35. Yu and coworkers demonstrated that a site‐selective Minisci‐type reaction [33] can be achieved using the electron‐poor O ‐acyl hydroxy‐amides 33as amidyl radical precursors and several N ‐heteroarenes 34( Scheme 2.9) [34]. Upon visible light irradiation, the excited dye 3CzCIIPN promoted SET reduction of 33(in analogy to the O ‐acyl oximes), thus enabling access to the corresponding nitrogen radicals 36. A 1,5‐HAT process gave the carbon radical 37, which underwent addition to an activated heteroarene to give 38. The authors suggested that under basic conditions, a proton and electron transfer took place, thus leading to the desired product 39. This reactivity displayed remarkable functional group tolerance in terms of the compatible heterocycles and was used for the modification of biologically active systems.
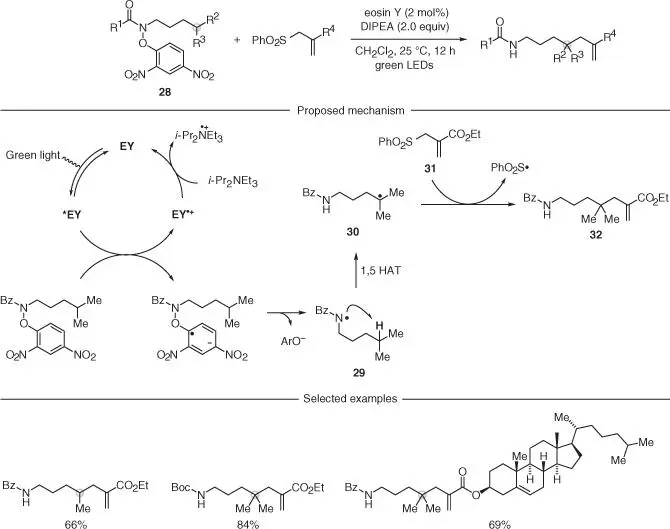
Scheme 2.8 Remote C(sp 3)–H allyation of amides using organic photocatalyst.
Source: Modified from Wu et al. [31].
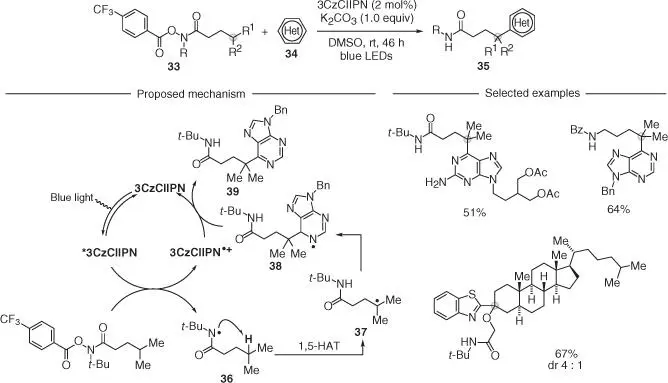
Scheme 2.9 Site‐selective remote C(sp 3)–H heteroarylation of amides.
Source: Modified from Chen et al. [34].
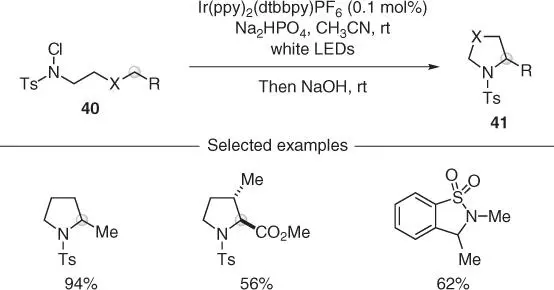
Scheme 2.10 Visible‐light‐promoted C(sp 3)–H amidation and chlorination of N ‐chlorosulfonamides.
Source: Modified from Quin and Yu [35].
The generation of amidyl radicals through photoinduced SET reduction is not restricted to electrophores embedded into a N–O systems but can also be performed on N ‐Cl derivatives. This was demonstrated by Yu and coworkers in the HLF‐type reactivity of N ‐Cl– N ‐Ts amines 40by using a heteroleptic Ir(III) photocatalyst under white light irradiation ( Scheme 2.10) [35]. In this case, after generation of a very electrophilic sulfamidyl radical, the 1,5‐HAT took place, and then the corresponding carbon radical was chlorinated, most likely, as part of a radical chain propagation. This protocol has also been used in the assembly of many pyrrolidines 41and also in late‐stage functionalizations of some bioactive materials, demonstrating the strong functional group compatibility.
2.3.2 Oxidative Strategies
2.3.2.1 1,5‐HAT via Iminyl Radicals
Inspired by the pioneering work of Forrester [17–20], the Leonori and the Studer group took carboxylic acid‐containing oximes 42and used them to generate functionalized imine and ketone derivatives 43–45( Scheme 2.11) [36–38]. These processes were all based on a reductive quenching photoredox cycle and required basic conditions to trigger the SET oxidation of the carboxylate functionality ( 46). This event led to two β‐scissions extruding CO 2and acetone and forming the corresponding iminyl radical 47. This species then underwent 1,5‐HAT and the incipient carbon radical 48was intercepted and diversified with several radical trapping agents (X–Y) such as N ‐chlorosuccinimide (NCS; chlorination, 43), selectfluor (fluorination, 44), and Michael acceptors (Giese addition, 45). A final electron transfer between the reduced photocatalyst and the Y· intermediate, generated upon atom transfer/addition, led to an overall redox‐neutral process. This approach for remote ketone functionalization tolerated important functionalities such as protected amines, heteroaromatics, free alcohols, and ester groups among others. Interestingly, in the case of NCS, the intermediate NH imine was directly intercepted by NCS before hydrolysis, leading to the corresponding N–Cl imines that showed remarkable stability.
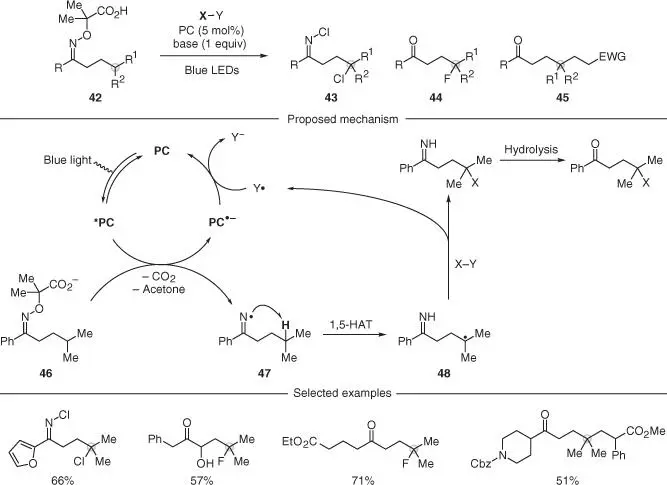
Scheme 2.11 Site‐specific intermolecular γ‐C(sp 3)–H functionalization of ketones via iminyl radicals.
Source: Jiang and Studer [36], Morcillo et al. [37], and Davies et al. [38].
2.3.2.2 1,5‐HAT via Amidyl and Sulfamidyl Radicals
A related approach for nitrogen radical generation was then used by the Leonori group to introduce a range of functionalities at the γ‐position of amides and at the δ‐position of protected amines using carboxylic acid‐containing hydroxyamides 49( Scheme 2.12) [37]. In analogy to their previous work on iminyl radicals, photoinduced oxidation of 49under basic conditions enabled amidyl radical generation, followed by 1,5‐HAT and distal carbon radical functionalization. In this case, halogenating agents such as NCS and selectfluor allowed introduction of γ chlorine and fluorine atoms, while S‐containing phthalimides enabled direct thioetherification. More importantly, 2‐iodoxybenzoic acid (IBX) reagents were successful to achieve remote C—C bond formation and introduce CN as well as alkyne functionalities. This strategy enabled the selective modification of tertiary, secondary, and even primary centers and was also used for the late‐stage functionalization of small dipeptides.
A very powerful approach for distal functionalization of amides has been possible from the corresponding N–H substrates using photoinduced proton‐coupled electron transfer (PCET). As initially demonstrated by Knowles and coworkers, this activation mode bypasses the need to install a stoichiometric electrophore on the substrates and therefore streamlines the preparation of the desired reaction products. This strategy has been initially used to trigger cyclization reactions [39], and also used in interrupted HLF‐type reactivity as demonstrated by the groups of Knowles and Rovis ( Scheme 2.13) [40, 41]. Following a reductive‐quenching photoredox cycle and in the presence of a suitable base, 50was effectively oxidized by photoinduced PCET generating the amidyl radical 51. This species was exploited in a 1,5‐HAT transposition and the resulting carbon radical 52reacted with a Michael acceptor ( 53), thus assembling a distal sp 3–sp 3C—C bond. Owing to electron‐poor nature of the intermediate α‐ester radical 53, the photoredox cycle was closed by SET with the reduced Ir(II) photocatalyst that delivered the stabilized anion 54that was protonated. The design of the amide substrates was critical to the feasibility and the overall success of the PCET as the carbonyl functionality must be able to acidify the N–H proton enough to encourage the deprotonation step. This somewhat controlled the nature of the amidyl radicals that could be generated. Nevertheless, this methodology displayed strong functional group tolerance as demonstrated by the number of Michael acceptors and functionalized amides that could be used.
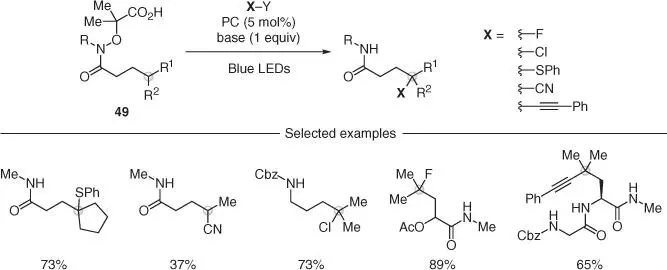
Scheme 2.12 Nitrogen radical mediated remote functionalization of amides and amines.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.