Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
2.3 Photoinduced Strategies
2.3.1 Reductive Strategies
2.3.1.1 1,5‐HAT via Iminyl Radicals
Iminyl radicals have been identified as powerful reactive intermediates for a number of reactions such as intramolecular cyclization but also radical ring opening and 1,5‐HAT. In this case, the pioneering work of Forrester [17–20] has clearly demonstrated their ability to partake into radical transposition, but the harsh chemical methods for their generation limited applications in mainstream organic synthesis.
The Leonori group recently reported that iminyl (and amidyl radicals) can be generated upon visible light irradiation from easy‐to‐make O ‐aryl oximes [21] via oxidative quenching photoredox cycles and also via the formation of electron donor–acceptor (EDA) complex with tertiary amines [22–24]. These strategies have been adopted by others in the subsequent development of interrupted HLF‐type reactions.
Fu and coworkers described a photochemical intramolecular α‐ N sp 3C–H imination based on O ‐aryl oximes 12for the preparation of functionalized imidazoles 13( Scheme 2.4) [25]. In this case, the authors exploited the known ability of these derivatives to undergo EDA complex formation with a tertiary amine in an intramolecular setting ( 12). This EDA complex underwent photoinduced single‐electron transfer (SET) and fragmentation across the weak N—O bond to give the iminyl 14. This slightly nucleophilic radical was then able to trigger a polarity‐matched 1,5‐HAT from the α‐N sp 3carbon providing 15. An ionic intramolecular cyclization, followed by aromatization, delivered the desired products 13. This methodology can only be used to assemble five‐membered ring systems and is limited on having an aryl substituent on the oxime. However, the site of 1,5‐HAT could be embedded into a further cyclic system, thus leading to interesting bicyclic structures.
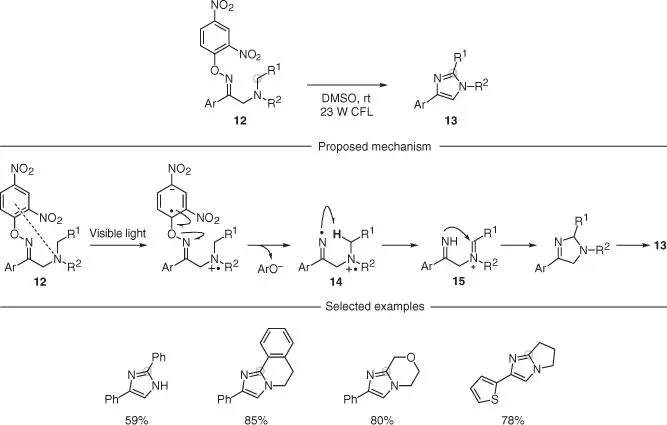
Scheme 2.4 Photochemical intramolecular C(sp 3)–H imination of tertiary aliphatic amines containing β‐ O ‐aryl oximes.
Source: Modified from Li et al. [25].
More recently, Wu and coworkers have demonstrated that similar O ‐aryl oximes 16can also undergo EDA complex formation with 1,4‐diazabicyclo[2.2.2]octane (DABCO)·(SO 2) 2 17and that this can be used for iminyl radical generation upon visible light irradiation ( Scheme 2.5) [26]. This step was followed by a 1,5‐HAT process, and the resulting carbon radical was able to intercept SO 2en route to products 18. In this way, a rare example of amino‐sulfonylation of unactivated sp 3C—H bonds was achieved, leading to the assembly of biologically relevant 1,2‐thiazine dioxides.
The ability of iminyl radicals to undergo 1,5‐HAT has been recently employed for the conversion of activated acyclic O ‐acyl oximes 19into a variety of cyclic ketones 20( Scheme 2.6). In this work, Nevado and coworkers described a redox‐neutral process, where the SET reduction of radical precursor 19was performed by a visible‐light‐excited Ir(III) photocatalyst [27]. In this case, the authors used O ‐acyl oximes that have a reduction potential accessible by many photoexcited catalysts [28]. This SET led to the corresponding iminyl radical 21that underwent selective 1,5‐HAT. The distal carbon radical 22was exploited in an intramolecular cyclization onto the (hetero)aromatic substituent. Oxidation of intermediate 23by the Ir(IV) catalyst gave the imine 24, which, upon hydrolysis, delivered a variety of fused bicyclic ketones in high yields. The six‐membered ring radical cyclization enabled excellent control of relative stereochemistry when substituents were present.
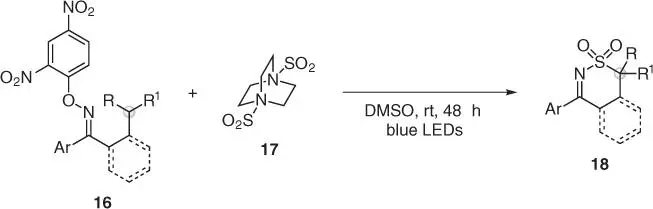
Scheme 2.5 Photoinduced aminosulfonylation of C(sp 3)—H bonds with sulfur dioxide.
Source: Modified from Li et al. [26].
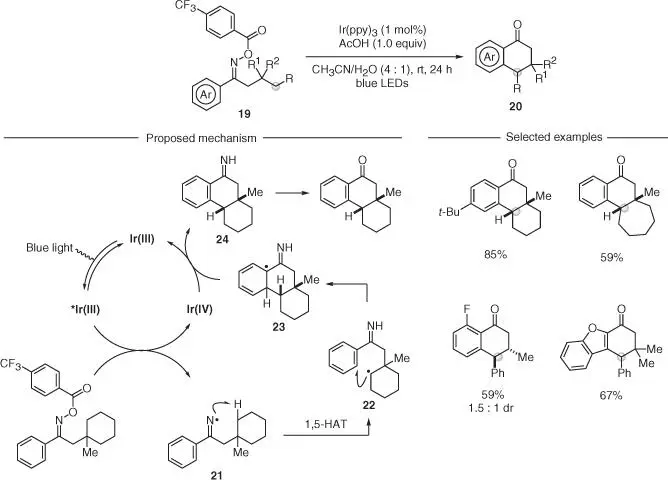
Scheme 2.6 Redox neutral remote functionalization of C(sp 3)—H bonds via iminyl radicals.
Source: Modified from Shu and Nevado [27].
The direct installation of vinyl groups onto unactivated sp 3C—H bonds is a transformation with powerful applications as the olefin functionality can then be elaborated in a number of different ways. An interesting solution to this challenge has been provided by Yu and coworkers that have developed cascade reactions between electron‐poor O ‐acyl oximes 25and styrenyl boronic acids 26( Scheme 2.7) [29]. This reactivity was also based on the generation of an iminyl radical by reductive SET from a photoexcited catalyst, its following transposition by 1,5‐HAT, and the reaction of the corresponding distal carbon radical with the olefin. Although the methodology was only used to install styrenyl‐type substituents, it enabled the fast assembly of small‐molecule building blocks 27difficult to target by other methodologies.
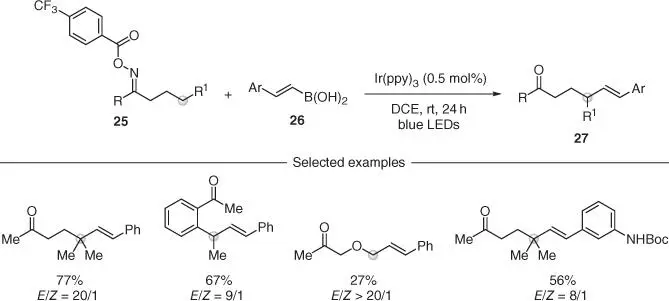
Scheme 2.7 Intermolecular remote C(sp 3)–H and C–C vinylations via iminyl radicals under photoredox‐catalysis.
Source: Modified from Shen et al. [29].
Shortly after, Duan and coworkers demonstrated that a related cascade reaction could be implemented using directly styrenes [30]. In this case, a broad range of substitution pattern was tolerated.
2.3.1.2 1,5‐HAT via Amidyl and Sulfamidyl Radicals
As discussed in Section 2.2, the strong electrophilic character of amidyl and sulfamidyl radicals means that very effective 1,5‐HAT transpositions can be achieved.
A powerful example based on SET reductions can be found in the work by Wang, who reported the remote sp 3C–H allylation of amides ( Scheme 2.8) [31]. In this case, the authors used the highly electron‐poor O ‐aryl hydroxyamides 28( 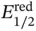 = −0.9 V vs. saturated calomel electrode [SCE]) that were previously identified as powerful electrophores for SET by the photoexcited state of the organic dye eosin Y (*
= −0.9 V vs. saturated calomel electrode [SCE]) that were previously identified as powerful electrophores for SET by the photoexcited state of the organic dye eosin Y (* 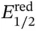 = −1.1 V vs. SCE). This event led to a fragmentation across the weak N—O bond, thus delivering the key amidyl radical 29. A very facile 1,5‐HAT process was used to generate the distal carbon radical 30, which underwent allylation by reaction with a variety of sulfone reagents (e.g. 31). This step provided the product 32with concomitant release of PhSO 2 ·. The authors proposed a redox‐neutral photoredox process, whereby N,N ‐diisopropylethylamine (DIPEA) closed the cycle by SET with the oxidized eosin Y. However, as quantum yield determination was not performed, a potential radical chain mechanism cannot be ruled out [32]. This reaction enabled the modification of secondary and tertiary centers and also displayed broad functional group compatibility.
= −1.1 V vs. SCE). This event led to a fragmentation across the weak N—O bond, thus delivering the key amidyl radical 29. A very facile 1,5‐HAT process was used to generate the distal carbon radical 30, which underwent allylation by reaction with a variety of sulfone reagents (e.g. 31). This step provided the product 32with concomitant release of PhSO 2 ·. The authors proposed a redox‐neutral photoredox process, whereby N,N ‐diisopropylethylamine (DIPEA) closed the cycle by SET with the oxidized eosin Y. However, as quantum yield determination was not performed, a potential radical chain mechanism cannot be ruled out [32]. This reaction enabled the modification of secondary and tertiary centers and also displayed broad functional group compatibility.
Интервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.