Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
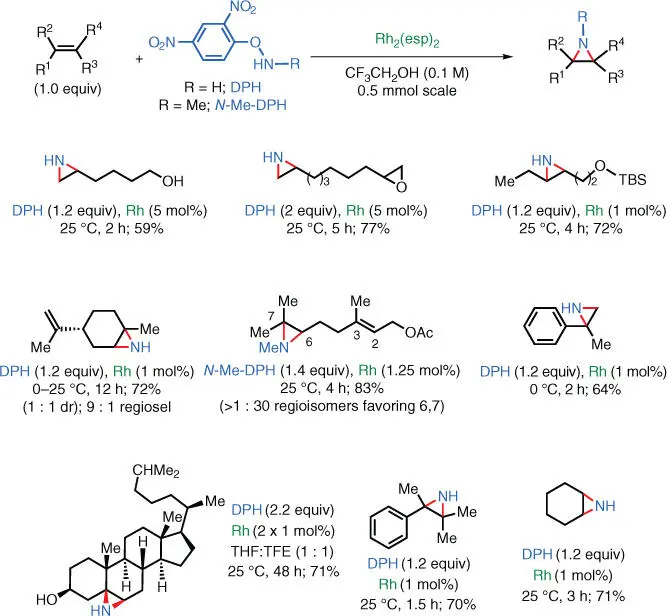
Scheme 1.29 Rh‐catalyzed NH‐aziridination of unactivated olefins using DPH.
Because N ‐alkyl HOSA derivatives can be readily prepared on multigram scale [46], it was demonstrated that N ‐Me as well as N ‐isopropyl units could be stereospecifically transferred to olefins, leading to the corresponding N ‐alkylaziridines.
Further studies on this catalytic system by the three groups showed that the Rh–nitrene intermediate can also facilitate the C–H amination of electron‐rich aromatic rings ( Scheme 1.31) [47]. In 2016, the Falck, Kürti, and Ess groups published the direct C–H amination of arenes. By using a more reactive aminating reagent and modifying the acidity of the reaction, Rh–nitrene intermediate can be directly inserted into the aromatic π‐system of electron‐rich arenes. Using this protocol, both inter‐ and intramolecular arene C–H amination can be achieved.
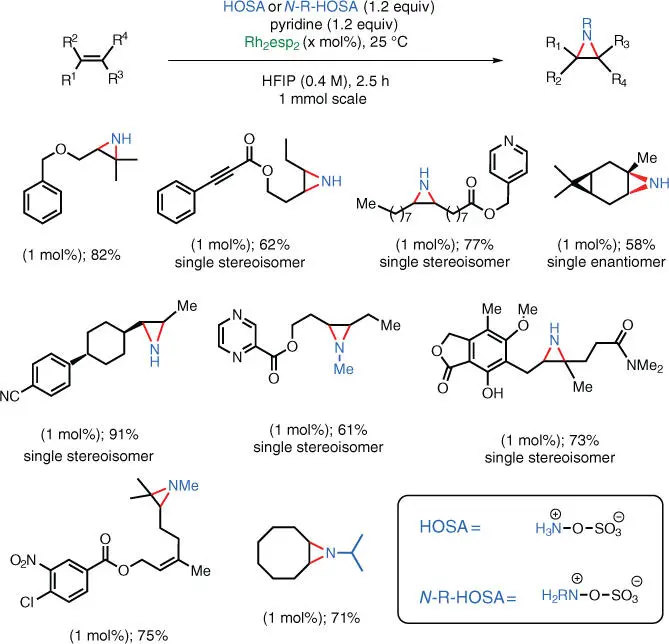
Scheme 1.30 Rh‐catalyzed NH‐aziridination of unactivated olefins using HOSA.
1.5 Transition‐Metal‐Free Electrophilic Amination Reactions
Although the mechanism of uncatalyzed electrophilic amination reactions seems straightforward, in practice, these uncatalyzed reactions often suffer from poor efficiency and low yields. A common issue with uncatalyzed electrophilic amination is the side reaction between the nucleophile and the highly reactive aminating reagent. It is especially difficult to directly synthesize primary or secondary amines under uncatalyzed conditions because the unmasked NH protons will usually quench the strongly basic nucleophiles used in the reactions ( Scheme 1.32).
In recent years, several approaches have been developed to address these issues and finally achieve practical TM‐free electrophilic aminations.
One approach is to use a mild nucleophile to avoid quenching and/or side reactions. In 2012, the Kürti group reported a TM‐free primary amination of arylboronic acids ( Scheme 1.33) [48].
This strategy exploits the fact that hydroxylamines can act as both electrophiles and nucleophiles. The nitrogen atom in the DPH aminating reagent first acts as a nucleophile to attack the boronic acid, and subsequently, it acts as an electrophile to accept an intramolecular nucleophilic attack to furnish the aniline products. Before this method, the direct conversion of arylboronic acids to the corresponding primary arylamines under mild conditions was not possible. Now, even halogenated primary arylamines may be readily prepared under transition‐metal‐free conditions.
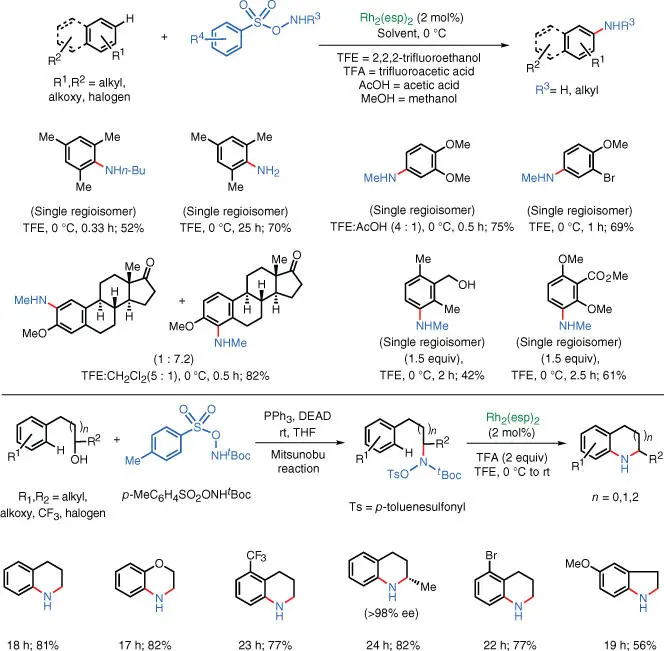
Scheme 1.31 Rh‐catalyzed aromatic C–H amination.
Source: Modified from Paudyal et al. [47].
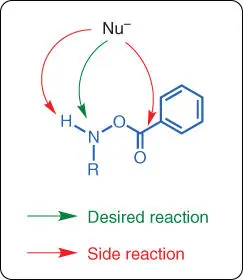
Scheme 1.32 Problems with uncatalyzed electrophilic amination.
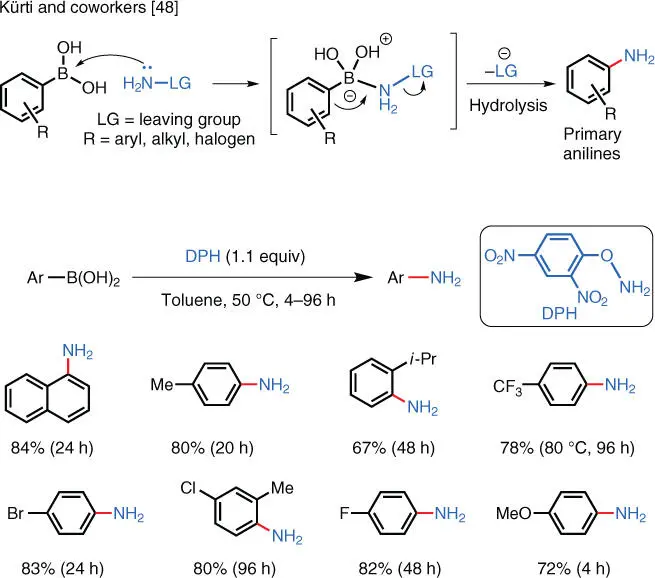
Scheme 1.33 TM‐free electrophilic amination of arylboronic acids.
Source: Modified from Zhu et al. [48].
Another approach to achieve TM‐free electrophilic amination while avoiding undesired quenching of the nucleophile is to modify the structure of the aminating reagent. In 2017, the Kürti group reported the use of sterically hindered oxaziridines to achieve the TM‐free direct primary amination of aryl Grignard reagents and aryl lithiums ( Scheme 1.34) [49]. Subsequent studies carried out in the Kürti group successfully expanded the substrate scope to include alkyl organometallic reagents [50].
The backbone of these aminating reagents can be readily recovered and reused for the preparation of more reagents (i.e. NH‐oxazirdines). Remarkably, the N—H bond does not undergo deprotonation as this pathway is sterically inhibited (i.e. because of the kinetic decrease of the N—H bond acidity). It is intriguing that the N ‐alkyl derivatives of these oxaziridines transfer the oxygen atom to aryl and alkyl Grignard reagents at low temperatures with complete chemoselectivity (i.e. no N‐transfer occurs with these reagents). Both the direct primary amination and hydroxylation of Grignard reagents are currently considered to be still extremely challenging, and they represent unmet synthetic needs. The camphor‐derived N ‐benzyl oxaziridine is significantly less reactive and considerably more stable than Davis's oxaziridine; thus, it allows for highly chemoselective hydroxylations in the presence of multiple sensitive functionalities such as sulfides, amines, and alkenes.
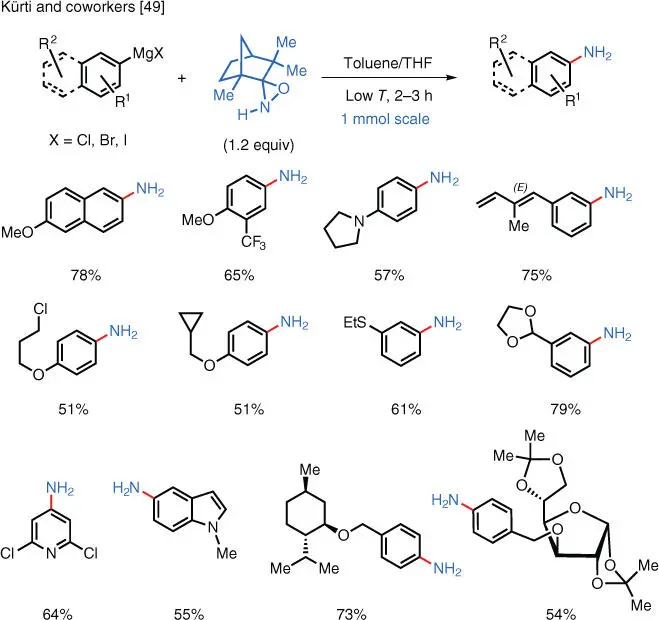
Scheme 1.34 TM‐free electrophilic amination of arylmetals using NH‐oxaziridines.
Source: Modified from Gao et al. [49].
For weaker nucleophiles such as olefins and arenes, successful TM‐free electrophilic aminations rely on highly reactive aminating reagents. Pioneering works in this area by the Bower group have shown that it is possible to achieve TM‐free intramolecular electrophilic amination using highly reactive sulfonyl hydroxylamines under acid catalysis ( Scheme 1.35). The unprotected tosylhydroxylamines are too unstable to be isolated, so they are instead generated in situ from the Boc‐protected precursors under acidic conditions. These reactions are proposed to proceed via the “Butterfly Mechanism” akin to the Prilezhaev reaction [51].
The α‐aminoketone moiety is commonly found in biological molecules, natural products, and active pharmaceutical ingredients. Despite their apparent importance, synthetic access to these compounds is not always straightforward. This is especially evident when the desired amino ketones have a primary amino group attached to a fully substituted α‐carbon atom. The most common strategy for synthesizing these compounds involves a two‐step approach, in which either an azido, a nitro, or a hydroxylamino group is first installed at the α‐position. A subsequent hydrogenation, usually catalyzed by a transition metal such as Pd, Pt, or Raney Ni, is then carried out to obtain the corresponding primary α‐aminoketone. Aside from the added steps, this approach also has limitations such as the lack of chemoselectivity during the reduction and the potential instability of the azido‐ or nitro‐intermediates.
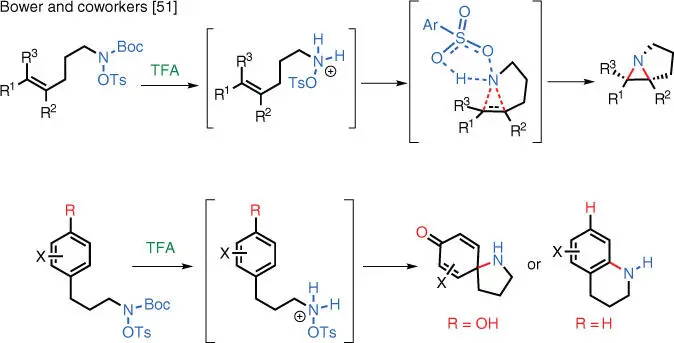
Scheme 1.35 TM‐free Prilezhaev reaction.
Source: Modified from Farndon et al. [51].
In 2019, the Kürti group reported the intermolecular TM‐free Aza‐Rubottom reaction [52]. Electron‐rich silyl enol ethers can be directly converted to the corresponding α‐primary amino ketones in one step. This reaction also proceeds via the “Butterfly Mechanism,” and the reactivity of the hydroxylamine aminating reagent is enhanced by cooperative hydrogen bonding interactions provided by the HFIP solvent molecules ( Scheme 1.36).
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.