Methodologies in Amine Synthesis
Здесь есть возможность читать онлайн «Methodologies in Amine Synthesis» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Methodologies in Amine Synthesis
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Methodologies in Amine Synthesis: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Methodologies in Amine Synthesis»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Methodologies in Amine Synthesis: Challenges and Applications Only up-to-date and comprehensive book on the preparation of amines ? one of the most frequently occurring compound classes found in natural products, bioactive molecules, and advanced materials. Presents efficient and useful synthetic methods, highlights opportunities / challenges as well as applications in pharmaceutical chemistry and materials science. Chapters are compiled by well-known experts in the field. One of them edited the previous books
(2001) and
(2007). The book
is a musthave for chemists in academia and industry working in the field of organic synthesis and catalysis, natural product chemistry, drug synthesis and pharmaceutical chemistry, as well as materials science.
Methodologies in Amine Synthesis — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Methodologies in Amine Synthesis», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
One important consideration in these types of reactions is that the nucleophile has to be able to coexist with the aminating reagent in order for the catalytic cycle to be established. Otherwise, the nucleophile may directly react (i.e. protonation and/or uncatalyzed nucleophilic attack) with the aminating reagent and may lead to the formation of undesired by‐products. This puts limitations on both the nucleophilicity and basicity of the organometallic nucleophiles. Organozinc compounds, Grignard reagents, and organoaluminum [12] reagents have been shown to be suitable nucleophiles for these types of reactions, while organolithium reagents are generally unsuitable because of their strong basicity and tendency to participate in side reactions that involve electron transfer pathways.
To address this issue and provide a solution for the direct electrophilic amination of organolithium reagents via copper catalysis, the group of A. B. Smith III (University of Pennsylvania, 2013) developed a protocol in which a siloxane transfer reagent is used to modulate the reactivity of the organolithium reagents ( Scheme 1.6) [13]. The siloxane reagent acts as an “attenuator” for the more reactive organolithium reagents and lowers its reactivity by forming a less basic complex that can more readily participate in the catalytic cycle and, in the meantime, prevents the direct attack on the aminating reagent by the organolithium.
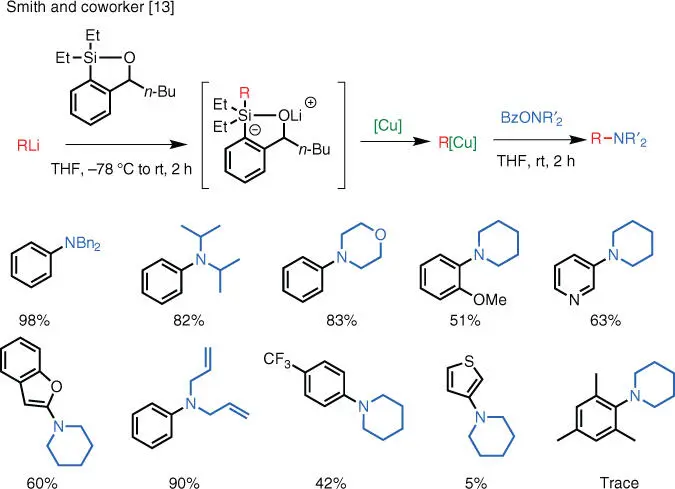
Scheme 1.6 Cu‐catalyzed electrophilic amination of organolithium reagents.
This type of C—N bond‐forming reaction can also take place directly between cuprates and electrophilic aminating reagents. One reason to justify the use of stoichiometric amounts of a copper salt (i.e. full transmetalation) is that the low basicity of the resulting cuprate can tolerate the presence of N–H protons in the aminating reagent, thereby enabling the synthesis of primary and secondary amine products without the use of excess organometallic reagent. The Kürti group has demonstrated this possibility with the use of sterically hindered N–H oxaziridines ( Scheme 1.7) [14]. Compared to the tertiary amines, unprotected primary amines are more versatile building blocks for further functionalization.
These sterically hindered N–H oxaziridines can be readily synthesized on multigram scale from the corresponding N–H imines and meta ‐chloroperbenzoic acid ( m CPBA). The N–H oxaziridines are bench‐stable compounds and can also be readily purified via flash column chromatography and using regular silica gel as the stationary phase. The steric hindrance created by the bulky alkyl groups reduces the kinetic acidity of the oxaziridine N—H bond, thus allowing the cuprates to be aminated as opposed to suffering unproductive proton transfer.
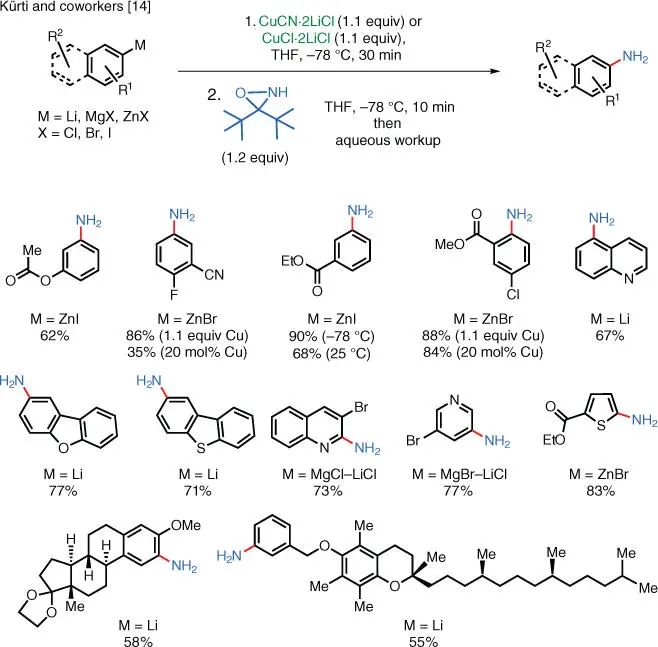
Scheme 1.7 Electrophilic amination of arylcuprates using a NH‐oxaziridine.
Since the advent of direct C–H cupration of arenes, it is now also possible to utilize cuprate nucleophiles without first going through a separate transmetalation step. Uchiyama and coworkers (at RIKEN, Japan) showed that it was indeed possible to directly aminate aryl cuprates with O ‐benzyl hydroxylamine. The directed C–H cupration of arenes was achieved using a strong base (TMP) 2Cu(CN)Li 2, which can selectively deprotonate at the ortho position of the amide directing group. The resulting aryl cuprates can be directly primary aminated with benzyl hydroxylamine ( Scheme 1.8) [15]. The hygroscopic nature of O ‐benzyl hydroxylamine necessitates its use as a stock solution in an organic solvent.
The instability of organometallic reagents used in the aforementioned reactions imposes limits on their widespread utilization, especially in an industrial setting. Efforts have been made to replace the air‐ and moisture‐sensitive organometallic reagents with more stable alternatives that can be conveniently stored and used. Miura and coworkers (Osaka University, Japan) have shown that both arylboronates ( Scheme 1.9) [16] and arylsilanes ( Scheme 1.10) [17] can serve as starting materials in Cu‐catalyzed electrophilic amination reactions. These reactions can proceed under ambient temperature and furnish the corresponding anilines in good to excellent isolated yields. The enhanced stability of arylboronates and arylsilanes reduces the complexity of the operation and, at the same time, the wide commercial availability of arylboronates also adds convenience to these types of reactions.
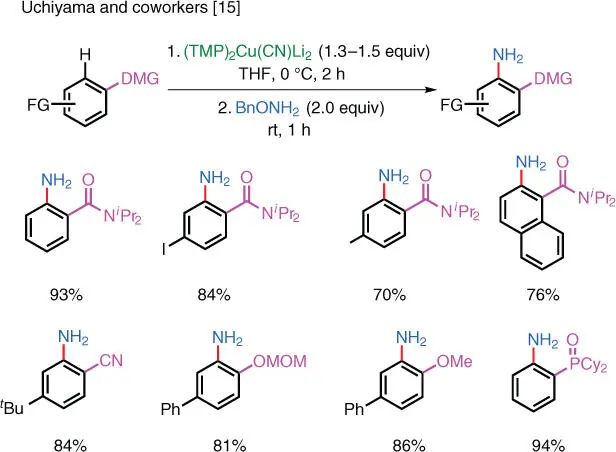
Scheme 1.8 Electrophilic amination via directed C–H cupration.
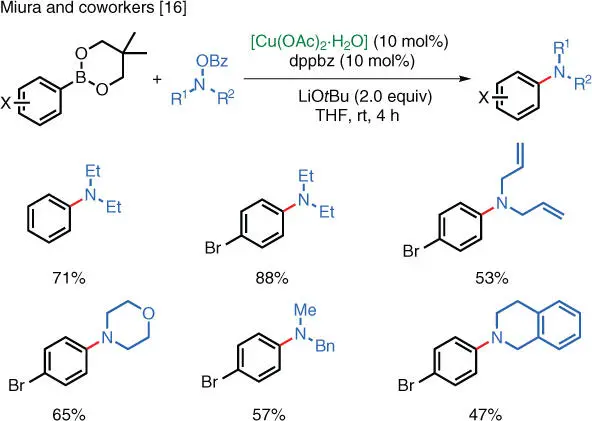
Scheme 1.9 Cu‐catalyzed electrophilic amination of arylboronates.
Hirano and Miura discovered that ambident nucleophiles such as silyl ketene acetals are also suitable nucleophiles for the Cu‐catalyzed electrophilic amination reactions. These reactions result in the formation of alpha‐amino esters as products. The first generation of this reaction uses chloramines as aminating reagents [18], while the second generation can proceed with the more stable and much safer O ‐benzoyl hydroxylamines ( Scheme 1.11) [19]. This method provides a potential route for the syntheses of unnatural as well as modified natural amino acids.
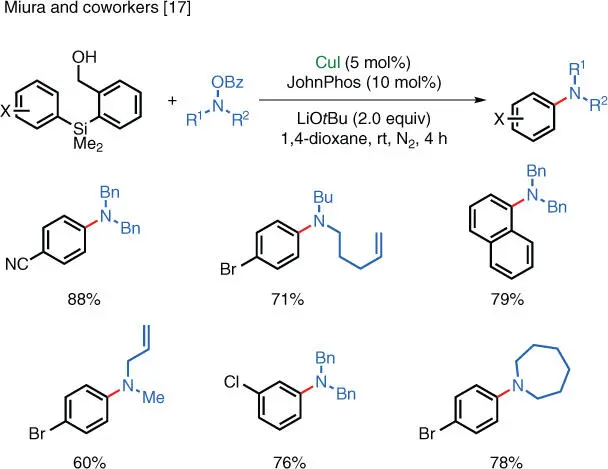
Scheme 1.10 Cu‐catalyzed electrophilic amination of aryl silanes.
Source: Modified from Miki et al. [17].
Weak nucleophiles such as styrenes and some electron‐deficient heterocycles can also participate in Cu‐catalyzed electrophilic amination reactions.
In the case of styrenes, the substrates can undergo hydroamination or aminoboration depending on the specific reaction conditions. Hirano, Miura, and coworkers have demonstrated that styrenes can be stereoselectively functionalized with benzoyl hydroxylamine and bis(pinacolato)diboron under Cu catalysis ( Scheme 1.12) [20]. The resulting products can further participate in transition‐metal‐catalyzed cross‐coupling reactions.
When polymethylhydrosiloxane (PMHS) is used instead of bis(pinacolato)diboron, hydroamination products can be obtained under similar reaction conditions ( Scheme 1.13). In these cases, it is proposed that the reaction proceeds with an initial CuH addition across the C—C bond of the olefins, followed by the electrophilic amination of the resulting cuprates [21].
With chiral ligands, the hydroamination reactions can give enantiomerically enriched products. Both the Miura ( Scheme 1.14) and Buchwald ( Scheme 1.15) groups have developed conditions using chiral phosphine ligands [21, 22].
Buchwald and coworkers have also reported the hydroamination of aryl acetylenes. The reaction is highly stereoselective, giving E ‐enamines as the major products. The enamine products can be further reduced to give alkyl amines, which are important building blocks in organic synthesis ( Scheme 1.16) [23].
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Methodologies in Amine Synthesis»
Представляем Вашему вниманию похожие книги на «Methodologies in Amine Synthesis» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.









Обсуждение, отзывы о книге «Methodologies in Amine Synthesis» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.