Patricia Yolanda Ciavarelli - Neurofibromatosis Tipo 1
Здесь есть возможность читать онлайн «Patricia Yolanda Ciavarelli - Neurofibromatosis Tipo 1» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на испанском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Neurofibromatosis Tipo 1
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Neurofibromatosis Tipo 1: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Neurofibromatosis Tipo 1»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Es un pequeño libro que muestra todas las formas de presentación de esta condición genética multifacética con manifestaciones a nivel de piel, ojos, huesos y predisposición a la formación de tumores en el sistema nervioso central y periférico. A pesar de presentar las típicas manchas café con leche en la piel desde la infancia, no logramos descender la edad de diagnóstico: razón por la cual insistimos en la difusión de esta rara entidad. Está dirigido a todos los profesionales de la salud con interés en esta forma de facomatosis: genetistas, clínicos, oftalmólogos, traumatólogos y neurocirujanos interesados en ampliar el conocimiento de la NF1, más allá de su especialidad y para los pacientes curiosos que siempre buscan información médica sobre su padecimiento.
Neurofibromatosis Tipo 1 — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Neurofibromatosis Tipo 1», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
La vía RAS/MAPK es crítica para el normal desarrollo y regulación de la proliferación, diferenciación, crecimiento, apoptosis y senescencia celular. La desregulación de esta vía causa desórdenes genéticos con características clínicas semejantes, denominados Rasopatías o Sindromes RAS/MAPK (8).
Estos desórdenes incluyen además de la neurofibromatosis tipo 1 (NF1) causada por haploinsuficiencia de neurofibromina; el síndrome de Legius (haploinsuficiencia de SPRED1); el síndrome de Noonan (NS) (mutaciones en PTPN11, SOS1, RAF1, KRAS, BRAF y NRAS); el síndrome de Leopard (mutaciones en PTPN11 y RAF1); el síndrome de Costello (mutaciones activadoras en HRAS); el síndrome cardiofaciocutáneo (CFC) (mutaciones en BRAF, MAP2K1 / 2 y KRAS); el síndrome similar a Noonan (mutaciones en SHOC220 o CBL) y otros.
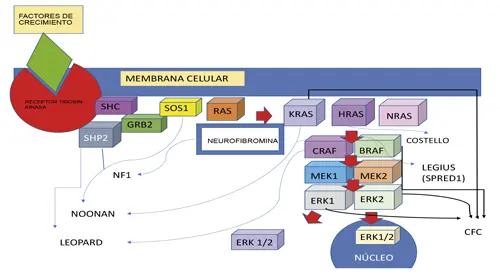
Fig. 5. La vía RAS/MAPK cuya alteración es responsable de múltiples sindromes (Rasopatías) como la neurofibromatosis.
Como ya se explicó, la neurofibrina actúa regulando negativamente a RAS, reduciendo la proliferación celular y diferenciación, previniendo la activación de la vía P13K/AKT/mTOR. En consecuencia, la pérdida de la función de NF1 aumenta los niveles de RAS-GTP y se activan todas las vías efectoras aumentando la proliferación celular y disminuyendo la apoptosis, como se comprueba en los neurofibromas y en los tumores malignos de nervio periférico.
La activación del RAS también activa la vía Raf-MAK/MEK (mitogen-activated-kinasa) para estimular al ERK (extracelular signal-relatedkinasa), la cual ingresa al núcleo celular y promueve la transcripción (9).
En síntesis, la variación patógena del gen NF1, afecta a la proteína neurofibromina que inhibe el crecimiento celular y de este modo queda liberada la cascada RAS con la producción de tumores (Fig. 6).
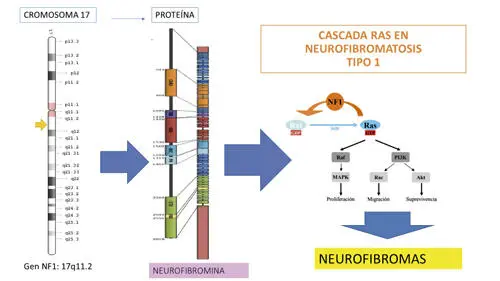
Fig. 6. Síntesis Gen-Proteína-Función
3. CRITERIOS DIAGNÓSTICOS DE LA NEUROFIBROMATOSIS TIPO 1
En 1987 el NIH (Instituto de Salud de EE. UU.) (10). definió los criterios en los que se basa el diagnóstico de la NF1 (revisados en 1997, actualmente en estudio por grupo de expertos), teniendo que cumplirse al menos 2 de los siguientes 7 criterios para llegar al diagnóstico de NF1:
• Seis ó más manchas café con leche (11). de > 5mm (prepuberal) o > 15 mm (postpuberal).
• Dos o más neurofibromas de cualquier tipo o 1 neurofibroma plexiforme.
• Pecas axilares o inguinales (signo de Crowe).
• Dos o más nódulos de Lisch (hamartomas del iris).
• Glioma del nervio óptico.
• Una lesión ósea característica, como displasia del esfenoides o adelgazamiento de la cortical ósea de los huesos largos con o sin pseudoartrosis.
• Un familiar de primer grado (hijo, hermano, padre o madre) con los criterios anteriores.
Vale la pena remarcar que estos criterios son muy sensibles y específicos en adultos; pero la aplicación en niños menores de 8 años puede variar. Además, no tienen en cuenta otras manifestaciones, como la dificultad de aprendizaje, los tumores malignos de la vaina nerviosa o la macrocefalia. Un estudio retrospectivo sobre 1.900 casos en el año 2000 demostró que casi el 50% de los casos esporádicos de NF1 no cumplían criterios diagnósticos antes del año de vida, mientras que a los 8 años, sí los cumplían el 97% y el 100% a los 20 años de edad.
En niñoslos criterios clínicos más comunes son las máculas (manchas) café con leche (95-100%), pecas en axilas o ingle (81%) y nódulos de Lisch (50-90%). Los Neurofibromas (15%), las lesiones óseas (60%) y los gliomas del óptico (15%), junto con el familiar de primer grado afectado completan los criterios.
Otras manifestaciones cardiovasculares, gastrointestinales y endócrinas pueden asociarse en la NF1.
4. MANIFESTACIONES CUTÁNEAS EN NF1
| Lesiones frecuentes | Lesiones infrecuentes |
| Manchas Café con leche | Xantogranuloma juvenil |
| Pecas o efélides | Glomus |
| Neurofibromas | Máculas azul-rojo |
| Aumento pigmentación | Melanoma |
| Máculas pseudoatróficas | |
| Nevus anémico |
4.1. Manchas Café con leche
La mácula café con leche (MCL) es la manifestación más temprana de la enfermedad (12). y uno de los siete criterios cardinales de diagnóstico. La lesión clásica es bien delimitada, con bordes lisos (“costa de California “) y homogénea en apariencia (Fig.7). En general, el color es cercana a la que su nombre indica; pero puede variar de marrón claro a marrón oscuro. Para cumplir con este requisito, los pacientes necesitan seis o más > 5 mm en prepúberes o > 15 mm en la post-pubertad. Menos del 1% de los niños menores de 5 años sin NF1 tienen más de dos; cuando son múltiples esto es altamente sugestivo de NF1 (13) (14) (15).
La prevalencia de la MCL en la población general ha variado entre 3 y 36% dependiendo de la población estudiada; pero la presencia de múltiples MCL en una población normal es generalmente menor a 1% (14).
La MCL es con frecuencia el primer signo, que se produce en el 99%de los pacientes con NF1 en el primer año de vida (14) (15). Continúan aumentando a lo largo de la infancia; pero a menudo se desvanecen en la edad adulta.
Las MCL obtienen su pigmento del melanocito, que se ha demostrado que tiene una mayor concentración de melanina y de melanosomas gigantes (macromelanosomas). Histológicamente las MCL presentan incremento de la melanina en los melanocitos y en los queratinocitos basales; pero sin proliferación melanocitaria.
A nivel de las organelas existe una mayor concentración de melanina y macromelanosomas tanto en la MCL como en la piel no MCL en comparación con controles sin NF1.
El melanocito en la MCL tiene una mutación en ambas copias del gen NF1 y los melanocitos de la piel no manchada de los pacientes con NF1, muestran sólo la mutación (16) de la línea germinal.
Las MCL no son patognomónicas de la enfermedad. Se calcula que hasta el 20% de los niños pueden presentar una MCL aislada, el 4% tienen 2 y menos del 1% de la población sana tiene más de 3, por lo que la anamnesis clínica y la exploración física deben ser particularmente rigurosas en este último caso. Lo primero que debemos valorar es si nos encontramos frente a una auténtica MCL, diferenciándola de otras lesiones pigmentadas como los nevus hipercrómicos (que pueden ser múltiples, pero tienen un borde mucho más irregular, Fig. 8), los mosaicismos pigmentarios, los nevus melanocíticos congénitos, los nevus spilus, la urticaria pigmentosa macular o la hiperpigmentación postinflamatoria.
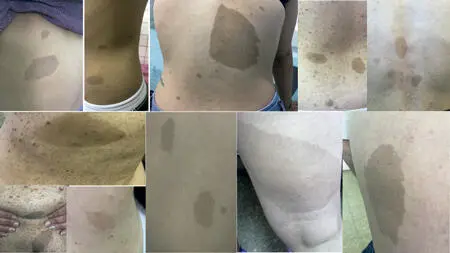
Fig 7. Manchas Café con Leche
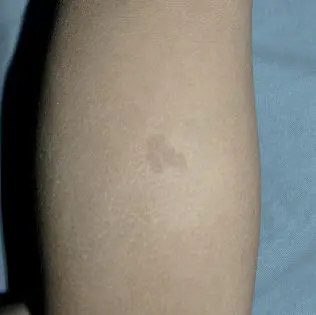
Fig 8. Nevus hipercrómico
El 99,3 % de nuestros pacientes (IC 95%: 97,3-99,9%), presentaron MCL.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Neurofibromatosis Tipo 1»
Представляем Вашему вниманию похожие книги на «Neurofibromatosis Tipo 1» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Neurofibromatosis Tipo 1» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.