Patricia Yolanda Ciavarelli - Neurofibromatosis Tipo 1
Здесь есть возможность читать онлайн «Patricia Yolanda Ciavarelli - Neurofibromatosis Tipo 1» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на испанском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Neurofibromatosis Tipo 1
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Neurofibromatosis Tipo 1: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Neurofibromatosis Tipo 1»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Es un pequeño libro que muestra todas las formas de presentación de esta condición genética multifacética con manifestaciones a nivel de piel, ojos, huesos y predisposición a la formación de tumores en el sistema nervioso central y periférico. A pesar de presentar las típicas manchas café con leche en la piel desde la infancia, no logramos descender la edad de diagnóstico: razón por la cual insistimos en la difusión de esta rara entidad. Está dirigido a todos los profesionales de la salud con interés en esta forma de facomatosis: genetistas, clínicos, oftalmólogos, traumatólogos y neurocirujanos interesados en ampliar el conocimiento de la NF1, más allá de su especialidad y para los pacientes curiosos que siempre buscan información médica sobre su padecimiento.
Neurofibromatosis Tipo 1 — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Neurofibromatosis Tipo 1», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
En 1940, Davis estableció el glioma óptico como parte de la patología. Glenn et al. en 1952, fueron los primeros en describir una paciente con neurofibromatosis e hipertensión arterial asociada con coartación de la aorta abdominal. Crowe, Schull y Neel, en 1956, investigaron los significados genéticos y clínicos de las máculas pigmentadas. Los estudios concluyeron que las manchas café con leche son signos patognomónicos de la neurofibromatosis.
Russel y Rubinstein en 1963 observaron dos tipos morfológicos de neoplasias: Neurofibromas y Schwannomas, que podrían encontrarse juntos dentro en una misma masa tumoral y que podrían presentar evidencias histológicas de malignidad.
Crowe en 1964 consideró las efélides axilarescomo un valioso signo para el diagnóstico de la neurofibromatosis.
Algunas manifestaciones clínicas inusuales fueron descritas por Diekinann et al. (1967), entre las cuales se encontraba la hipertensión arterialdebido a la estenosis de la arteria renal. La participación del corazón en la neurofibromatosis fue descripta y revisada por Rosenquist (1970), que también estudió el compromiso de la aorta abdominal, renal, carótidas y otras arterias.
Westerhof et al. (1983) relacionaron el hipertelorismocon la enfermedad y lo hallaron en el 24% de los pacientes con neurofibromatosis. Homwich et al. (1983) presentaron evidencias de pacientes con estenosis del acueducto de Silvioy neurofibromatosis. Crawford (1986), en estudio con 116 pacientes menores de 12 años, describió la presencia inusual de una paciente, con rabdomiosarcomaafectando el tracto genitourinario, que presentaba también una pseudo-artrosis congénita de la tibia. Crawford afirmó que la mayoría de los rabdomiosarcomas asociados a la neurofibromatosis involucran el tracto genitourinario. En 9 casos de neurofibromatosis hallaron tumores carcinoidessegún Griffiths et al. (1987), todos se localizaron en el duodeno y eran histológicamente distintos con inmunorreatividad para somatostatina.
Senveli et al. (1989) presentaron 6 pacientes con NF1 con estenosis de acueducto e hidrocefalia. La edad de los pacientes variaba entre 4 y 24 años. Veinte casos similares ya se habían presentado en la literatura en ese momento.
Wright et al. (1989) fueron los primeros en estudiar la historia natural del glioma del ópticoen niños con NF1, afirmando que éste podría tener un curso más benigno que los tumores esporádicos, Jadayel et al. (1990) utilizaron métodos moleculares para identificar el origen, en los padres, de las nuevas mutaciones de NF1. En 12 de 14 familias analizadas, se encontró que la nueva mutación era de origen paterno, aunque existían pocas o ninguna evidencia de los efectos de la edad paterna en la incidencia de las mutaciones.
Ragge et al. (1993) realizaron un estudio sobre los nódulos de Lisch, acompañado por fotografías del iris de diferentes colores. Sakurai ya había publicado 2 años antes que Lisch, una ilustración con las características de los nódulos del iris de los pacientes con Neurofibromatosis. Algunos postulan que deberían denominarse Nódulos de Sakurai-Lisch.
El carácter familiarquedó bien demostrado por Stojanovic et al (1993), que describieron un caso de neurofibromatosis generalizado en un joven de 21 años de edad y en su padre de 59 años. Su concomitancia con patología maligna fue enfatizada por Ferreira et al. (1994) en un caso de osteosarcoma asociado a Neurofibromatosis.
Legius et al. (1994) estudiaron el perfil neuropsiquiátricode 46 niños con una reducción en el lQ total, con un índice verbal mejor que en los otros grupos. La dificultad en la concentración fue especialmente significativa en los niños con alto lQ. Legius et al. (1994) sugirieron que estos niños podrían beneficiarse con el uso de ritalina.
Nopajaroonsie Lurie (1996) describió la presencia de un aneurisma venoso, displasia arterial y hemorragiaen un paciente de 62 años de edad. Este paciente presentaba un aneurisma en la vena yugular interna, que estaba asociado a displasia de arterias cervicales. Se encontraron tejidos neurofibromatosos en la pared del aneurisma, así como en las venas de menor calibre. Durante la cirugía del aneurisma, el paciente desarrolló una hemorragia masiva que fue dificultoso su control por la gran fragilidad de los vasos sanguíneos. Yamauchi et al. (2000) relataron la existencia de más de 50 casos de asociación entre NF1 y la enfermedad de Moyamoya.
2. NOCIONES BÁSICAS DE GENÉTICA
2.1 Estructura génica
En 1987 se localizó el gen causante de la neurofibromatosis mediante técnicas de clonación posicional en la región pericentromérica del cromosoma 17, en la zona 17q11.2 (Fig. 4).
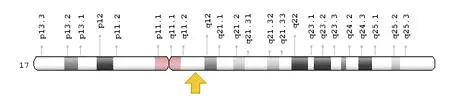
Fig 4. El gen NF1 fue identificado en 1990, de acuerdo a GRCh38.p12 (2) .
2.2 Genética molecular
Realizaremos un breve análisis de las diferentes variantes patógenas (antes denominadas mutaciones) que inactivan al gen de la NF1.
La NF1 presenta una alta tasa de mutaciones, ocurre una cada 10.000 gametas por generación (100 veces mayor que cualquier otro gen). Esta elevada tasa de mutaciones es atribuída a su gran tamaño. Más del 50% de los pacientes no tienen antecedentes familiares o sea que presentan mutaciones “de novo”. La penetrancia es completa luego de la infancia y su variabilidad clínica es muy alta no sólo entre individuos no relacionados; sino también entre individuos afectados dentro de una misma familia (portadores de la misma mutación) y en una misma persona en diferentes momentos de su vida. Esto sugiere que existen factores modificadores o epigenéticos responsables de la variabilidad de la expresión fenotípica de NF1 (3). Identificar las alteraciones que causan este síndrome resulta un desafío complejo debido al gran tamaño del gen, a la ausencia de puntos con alta probabilidad de mutaciones (hotspots) y a la existencia de pseudogenes (4). Aún así, se han descripto más de 1200 mutaciones diferentes (5).
La mayoría de las alteraciones (~ 85-90%) son mutaciones puntuales: sustituciones, inserciones y deleciones. Otras alteraciones son deleciones o duplicaciones de uno o varios exones (~2%) y microdeleciones (~ 5-10%) que abarcan el gen NF1 y genes vecinos (6).
Las alteraciones puntuales y las deleciones/duplicaciones pequeñas dan lugar a una proteína trunca originada por mutaciones sin sentido (non sense), pequeñas deleciones /inserciones con cambio en el marco de lectura (frame shift) y del sitio de splicing (splicesite). También se han descripto variantes patógenas (ex mutaciones) con cambio de sentido (missense) que producen alteraciones en la función de la neurofibromina.
Como se dijo anteriormente las alteraciones del gen NF1 dan lugar a un fenotipo extremadamente variable, motivo por el cual resulta aún hoy muy difícil establecer correlaciones genotipo- fenotipo (7) (algunas ya han sido descriptas).
2.3 Función de la neurofibromina
La neurofibromina regula negativamente la actividad Ras, favoreciendo la forma inactiva –Ras unido a GDP- en detrimento de la forma activa –Ras unido a GTP- (Figura 5). Por lo tanto, la existencia de mutaciones en NF1 que den lugar a alteraciones estructurales o ausencia de neurofibromina producen un aumento constante de la forma GTP- Ras, acelerando diversas cascadas de señalización que llevan a un aumento de la supervivencia, la proliferación y migración celular.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Neurofibromatosis Tipo 1»
Представляем Вашему вниманию похожие книги на «Neurofibromatosis Tipo 1» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Neurofibromatosis Tipo 1» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.