Masahiro Irie - Diarylethene Molecular Photoswitches
Здесь есть возможность читать онлайн «Masahiro Irie - Diarylethene Molecular Photoswitches» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Diarylethene Molecular Photoswitches
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Diarylethene Molecular Photoswitches: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Diarylethene Molecular Photoswitches»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Diarylethene Molecular Photoswitches — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Diarylethene Molecular Photoswitches», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Table 2.1 Relative ground state energy differences between the open‐ and closed‐ring isomers.
| Compound | Disrotatory (kJ/mol) | Conrotatory (kJ/mol) |
|---|---|---|
| 1,2‐Diphenylethene ( 9) | 175 | 114 |
| 1,2‐Di(3‐pyrrolyl)ethene ( 10) | 135 | 65 |
| 1,2‐Di(3‐furyl)ethene ( 11) | 113 | 38 |
| 1,2‐Di(3‐thienyl)ethene ( 12) | 51 | −14 |
On the contrary, orbital symmetry forbids the conrotatory cyclizations in the ground states from 9oto 9cand from 11oto 11c, because each S 0open‐ring isomer state correlates with a highly excited state of the closed‐ring isomer, as shown in Figure 2.2. On the other hand, no such large barrier exists in the S 1state for 9oand the S 2state for 11o. This indicates that electrocyclic reactions of both 1,2‐diphenylethene and 1,2‐bis(3‐furul)ethene are allowed in the photochemically excited states.
What should be discussed here is the stability of the closed‐ring isomers. Figure 2.2shows that in both 9cand 11c, the cycloreversion reactions in the ground state have to overcome energy barriers, and the barriers correlate with ground state energy differences between the open‐ and closed‐ring isomers. The calculated energy differences are shown in Table 2.1. When the energy difference is large, as in the case of 9, the energy barrier becomes small and the cycloreversion reaction takes place readily. On the other hand, the energy barrier becomes large when the energy difference is small. In this case, the cycloreversion reaction hardly takes place. The correlation between the ground state energy difference and the energy barrier is well explained by the Horiuti–Polanyi rule as shown in Figure 2.3. The energy difference in the ground states between the open‐ and closed‐ring isomers controls the stability of the closed‐ring isomers.
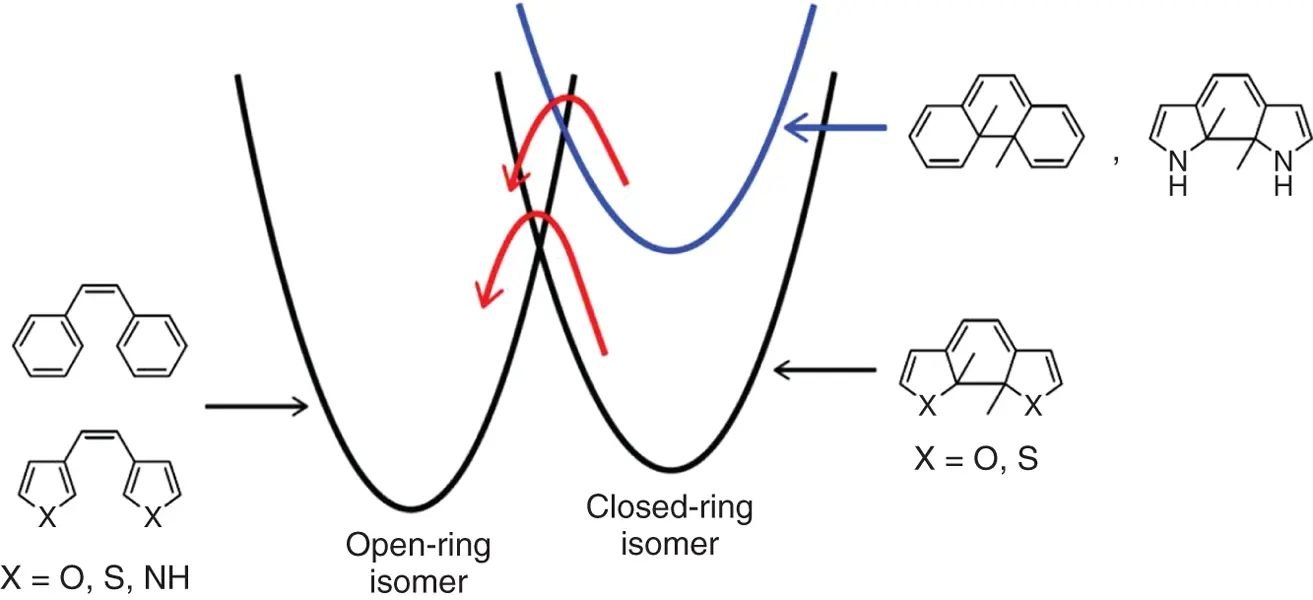
Figure 2.3 Correlation between the ground state energy difference between open‐ and closed‐ring isomers and the energy barrier.
The next question is what causes the difference in the ground state energy levels of the two isomers. First, strain energies of the six‐membered rings of the closed‐ring isomers were compared. The optimized geometries of the closed‐ring isomers, 9cand 11c, however, showed almost identical six‐membered ring structures and the ring‐strain could not explain the energy difference. Next, the aromaticity change from the open‐ to the closed‐ring isomers was examined. During the cyclization reaction, phenyl and heterocyclic rings change the structures as shown in Scheme 2.3. The aromaticity of the rings is lost during the cyclization reactions. The energy differences between the right‐ and left‐side groups were calculated and are shown in Table 2.2. The aromatic stabilization energy of the aryl groups correlates well with the ground state energy difference. The highest energy difference was calculated for the phenyl group and the lowest one for the thienyl group. Destabilization due to destruction of the aromatic ring during the cyclization reaction increases the energy of the closed‐ring form. The aromaticity is the key molecular property that controls the thermal stability of the closed‐ring isomers.
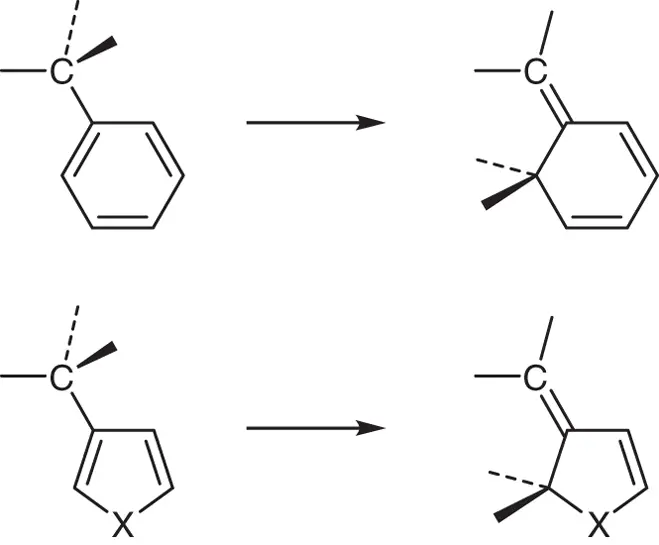
Scheme 2.3 The structure changes of phenyl and five‐membered heterocyclic rings along with the cyclization reactions.
Table 2.2 Aromatic stabilization energy differences.
| Group | Energy (kJ/mol) |
|---|---|
| Phenyl | 116 |
| Pyrrol | 58 |
| Furyl | 38 |
| Thienyl | 20 |
The theoretical prediction was confirmed by the synthesis of diarylethene derivatives with various types of aryl groups as shown in Figure 2.4. When the aryl groups are thiophene, benzothiophene, thiazole, or oxazole rings, which have low aromatic stabilization energy, the closed‐ring isomers are stable (more than 12 hours at 80 °C). On the other hand, photogenerated closed‐ring isomers of diarylethenes with indole rings, which have intermediate aromatic stabilization energy, undergo thermally reversible photoswitching (half‐life time at 80 °C of 16c: 2.5 hours). The closed‐ring isomers of diarylethene derivatives with phenyl rings readily returned back to open‐ring isomers (half‐life time at 20 °C of 18c: 1.5 minutes).
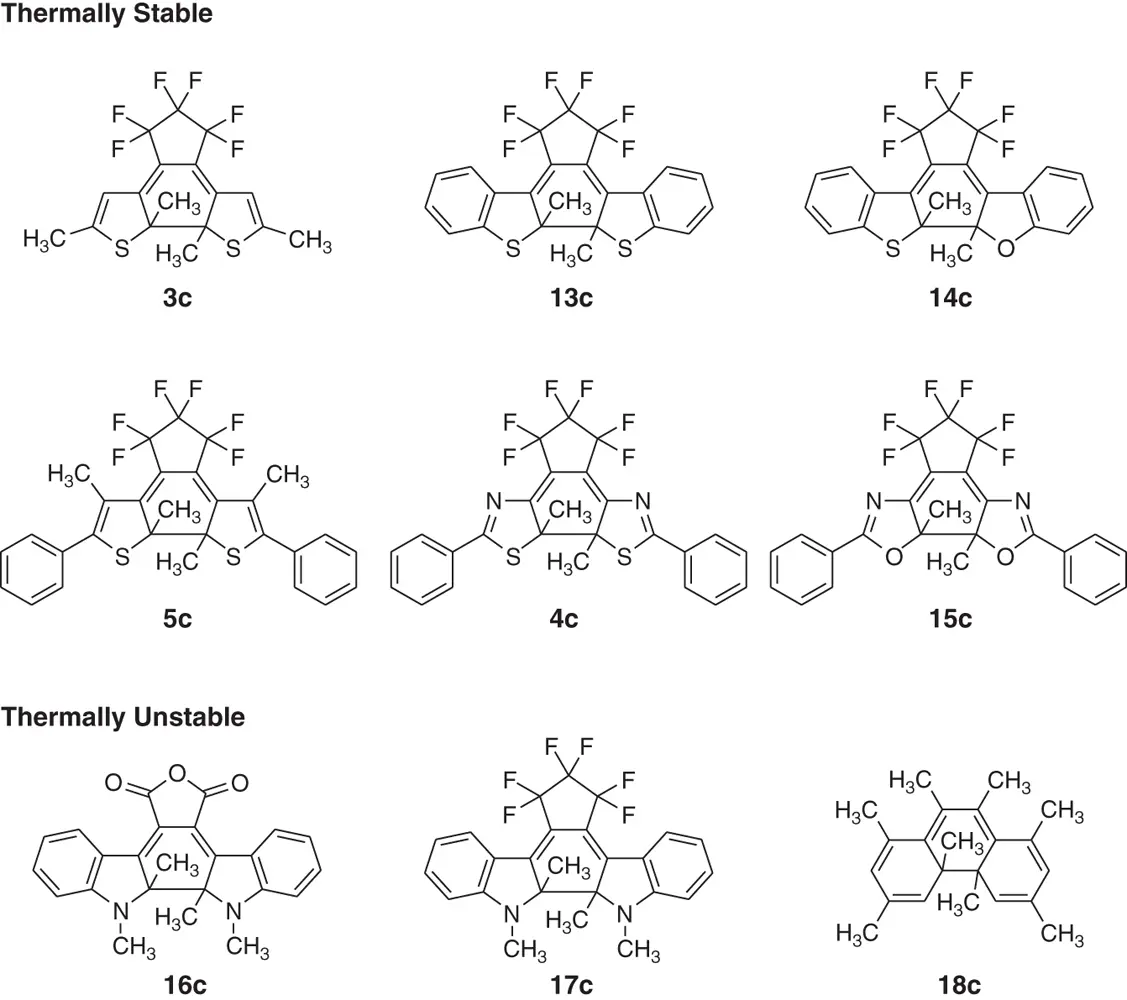
Figure 2.4 Thermal stability of diarylethene derivatives. Any appreciable change of the absorption intensity of the closed‐ring isomer was not observed in the thermally stable derivatives for more than 12 hours at 80 °C.
From the above theoretical and experimental results, the guiding principle for the synthesis of thermally irreversible diarylethenes is defined as follows.
The thermally irreversible photoswitching diarylethenes can be prepared by employing aryl groups with low aromatic stabilization energy.
2.2 Theoretical Study
The well‐studied photoinduced cyclization and cycloreversion reactions between 1,3,5‐hexatriene (HT) and cyclohexadiene (CHD) provide a useful framework for understanding the basic reaction mechanism of diarylethenes. As the first step, potential energy surfaces of a model diarylethene, 1,2‐bis(cyclopenta‐1,3‐dien‐2‐yl)‐ethene, were calculated using a complete active space self‐consistent field (CASSCF) method [3]. Figure 2.5shows S 0and S 1potential energy surfaces along the reaction coordinate. On both S 0and S 1surfaces there exist two minima, a closed‐ring isomer (CHD and CHD*) and an open‐ring isomer (HT and HT*). Transition structures (TS 0and TS 1) were also characterized on each potential energy surface. Several conical intersection minima (indicated by crosses) exist on both closed‐ and open‐ring sides of the potential surface in addition to CI 3. CI 3is the most important conical intersection, because it is the only one that provides a pathway toward both open and closed minima on the ground state. At the conical intersection geometry, one has a triangular arrangement between three unpaired electrons belonging to carbon atoms in the two 5‐membered rings. These three electrons are weakly coupled π‐electrons, and a fourth (belonging to a three‐electron allyl fragment) is an uncoupled spectator. It is worth noting that the TS 1barrier does not exist in the CHD/HT system. The barrier is probably due to steric constraint from the sigma‐bond framework.
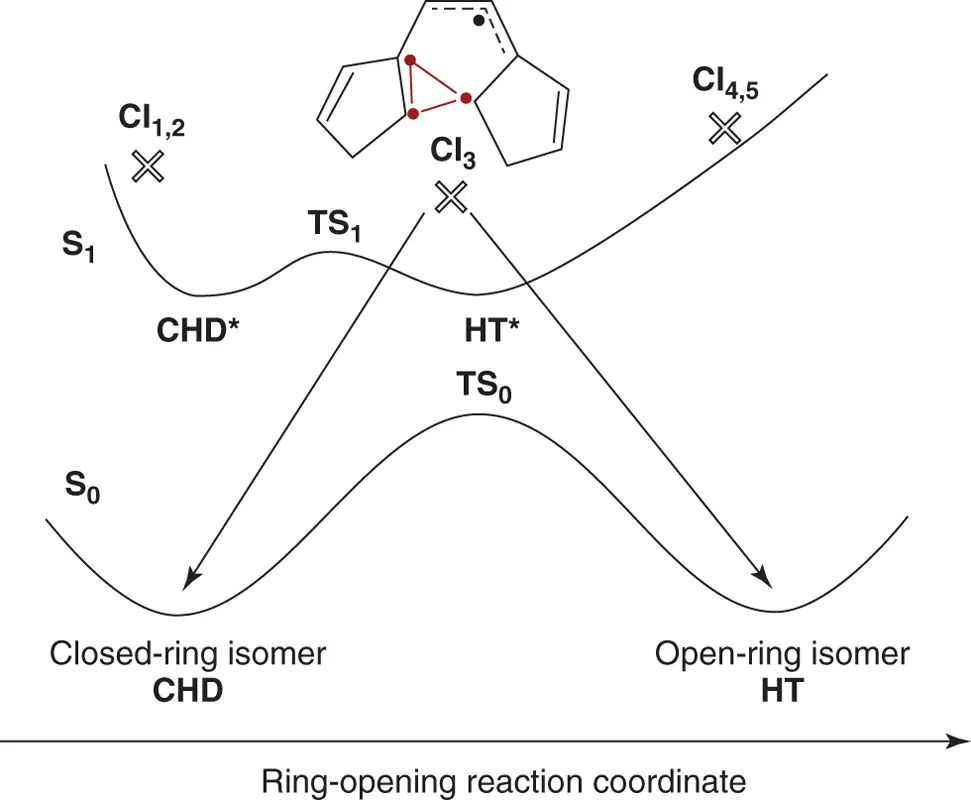
Figure 2.5 Potential energy surfaces of a model diarylethene.
Source : Reprinted with permission from Ref. [3]. Copyright 2003 American Chemical Society.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Diarylethene Molecular Photoswitches»
Представляем Вашему вниманию похожие книги на «Diarylethene Molecular Photoswitches» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Diarylethene Molecular Photoswitches» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.