Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
Здесь есть возможность читать онлайн «Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
The first book dedicated to the emerging field of physiologically based pharmacokinetic modeling (PBPK) Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulations: Principles, Methods, and Applications in the Pharma Industry
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry, Second Edition
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Determination of IC 50 shift, maximum inactivation rate (k inact ), and inactivator concentration at half‐maximal k inact (K I ) for TDI:The clinical value of in vitro models that address TDI of drug‐metabolizing enzymes has been reviewed (Venkatakrishnan et al., 2005; Grimm et al., 2009). IC 50shift (Obach et al., 2007) and projected IC 50(Atkinson et al., 2005) assays are generally the first screening methods to assess TDI. Determination of the kinetic parameters k inactand K I(Atkinson et al., 2005; Obach et al., 2007) is often carried out for promising drug candidates. The test compound is preincubated with an enzyme source (usually pooled human liver microsomes) in at least five different inhibitor concentrations to get the pseudo first‐order rate constant ( k obs), which is related to k inactand K Iby the Michaelis–Menten equation:
(2.1) 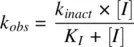
k inactand K Ican be obtained either by a nonlinear regression of equation 2.1or by plotting the Kitz–Wilson plot (1/ k obsvs. 1/[ I ]). Different in vitro approaches to evaluate MBI have been reviewed (Grime et al., 2009). The quasi‐irreversible MIC generated by coordinate bond formation to iron in the heme is identifiable by the absorbance of Soret peak at 450–455 nm by difference spectra scanning (Franklin, 1991) in a dual‐beam spectrophotometer. Human liver microsomes or recombinant enzyme can be incubated with the test compound. Nitrogen‐based MICs are further characterized by the reduction of the Soret absorbance on oxidation by potassium ferricyanide (Watanabe et al., 2007; Grimm et al., 2009). A key parameter that is necessary for the evaluation of TDI is the CYP degradation rate constant ( k deg) for the isoform that is inactivated. However, measured values vary widely (Yang et al., 2008). Intestinal inhibition needs to be considered for the TDI observed for CYP3A4 enzyme (Galetin et al., 2006).
Determination of E max and IC 50 for CYP induction:Any in vitro system employed to evaluate induction potential of a candidate drug should retain activity for a considerable length of time. In vitro systems employed to study enzyme induction are cultured human hepatocytes (LeCluyse et al., 2000; Kato et al., 2005; Luo et al., 2005), immortalized hepatocytes (Mills et al., 2004), minimally derived cell lines (Aninat et al., 2006) like HepaRG cells (Kanebratt and Andersson, 2008), reporter gene assays (El‐Sankary et al., 2001; Persson et al., 2006; Sinz et al., 2006), and ligand binding assays. The classic method that is widely used in the pharmaceutical industry and accepted by the FDA to measure induction is incubation of test compound with cultured human hepatocytes in which changes in mRNA expression and/or enzyme activity of target genes are measured and compared to untreated control hepatocytes (Chu et al., 2009). Advantages of hepatocytes are many. They contain native receptors and transporters; target genes are in their native context with full complement of regulatory elements. The disadvantage comes from the interdonor variability in CYP levels, which is difficult to distinguish from actual variability in induction response across human population. Methods using in vitro data to predict induction potential have been reviewed (Fahmi and Ripp, 2010). Focusing on CYP3A induction, an Innovation and Quality (IQ) Induction Working Group (WG) used a large clinical dataset to highlight the variability in in vitro parameters due differences in protocols, reagents, donors, analysis (methods and instrumentation) and to recommend in vitro evaluation of induction in three separate human donors (Kenny et al. 2018). If unbound maximum plasma concentrations or dose of the perpetrator are not known, the highest concentration tested in vitro is limited by aqueous solubility or cytotoxicity.
2.4.2 Candidate Drug as a Potential Victim of Inhibition
To evaluate a compound’s potential to be a victim of DDI, it is necessary to identify the specific enzymes involved in its metabolism. A common experimental approach to reaction phenotyping(Harper and Brassil, 2008; Zhang et al., 2009) is the use of cDNA‐expressed recombinant enzyme systems, in which the test compound is incubated with a panel of individually expressed human recombinant enzymes. A typical panel of CYP enzymes includes CYPs 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, and 3A5. Using the known CYP abundances 60, 61, the percentage contribution of individual CYPs to the overall oxidative metabolism of a drug candidate ( f m,CYP) can be estimated:
(2.2) 
CL int,iis the intrinsic clearance of the i th CYP isoform in individually expressed recombinant enzyme. The application of this method to estimate the relative contributions of the individual UGT enzymes to the overall glucuronidation rate is not so feasible today as the relative abundances of the various UGT enzyme activities in human liver are not well‐established. Altenatively, reaction phenotyping experiments are also done by incubating the test compound in hepatocytes, microsomes, or some other in vitro preparation, using normal tissues as the enzyme source with inclusion of selective chemical or immunoinhibitors of specific enzymatic pathways. By performing a series of incubations with various inhibitors, and comparing the relative rates of metabolism, one can identify which inhibitor reduces the overall metabolism to the greatest extent and thereby uncover the metabolic pathway that contributes the most to the clearance of a compound. The use of hepatocytes guarantees the full range of phase I and phase II enzymes but is limited by the availability of specific inhibitors to some enzymes (example, UGT enzymes). The percentage contribution of an enzyme isoform to the metabolism of a victim drug provides a measure of the extent of its dependence on that isoform ( f m,CYP). In addition, a good assessment of victim potential requires knowledge of the fractions eliminated in bile and urine in order to get the fraction of compound metabolized ( f m).
2.5 SOURCES OF UNCERTAINTY
In vitro data are associated with uncertainty due to knowledge gaps in system parameters or scalars. The data may also vary considerably between laboratories due to differences in donors and assays. When an NCE is a perpetrator of drug interaction, the unbound drug concentration at the site of interaction is uncertain, if the compound is a transporter substrate and/or highly bound to plasma proteins. This is especially true for efflux transporter inhibition or for CYP induction, where the driving concentrations are intracellular drug concentrations. The uncertainty in driving concentrations are even higher when these processes occur in the gut. In early development, the therapeutic dose of the perpetrator NCE drug is not identified and can add to the uncertainty in the DDI risk assessment. Sources of uncertainty are listed below:
NCE as victim
Contribution of gut for CYP3A and UGT substrates, fg;
Fraction metabolized, fm
fm,CYP
NCE as perpetrator
In vitro interaction parameters (Ki, kinact, KI, EC50 )
In vivo relevance of transporters in determining the intracellular concentrations of inhibitors that are substrates for uptake and/or efflux transporters
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»
Представляем Вашему вниманию похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.