Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
Здесь есть возможность читать онлайн «Sheila Annie Peters - Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
The first book dedicated to the emerging field of physiologically based pharmacokinetic modeling (PBPK) Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulations: Principles, Methods, and Applications in the Pharma Industry
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry, Second Edition
Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Among the efflux transporters, interactions with P‐gp are the most studied. Inhibition of P‐gp may not result in clinically significant differences in the exposure of the victim drug, but it can attenuate the efficacy of drugs targeting barrier tissues such as brain, lymphocytes, and tumor.
2.3 FACTORS AFFECTING DDI
The extent of DDI will depend on the characteristics of the perpetrator and victim drugs involved in the interaction. Perpetrator and victim properties impacting drug interaction risk are summarized in Figure 2.2. For an inhibitor, the greater the unbound inhibitor concentration relative to its potency, the greater is the extent of inhibition. Inhibitor concentrations at any point of time will depend on the dose administered and the organ where the affected enzyme or transporter is located. For a victim of inhibition, the greater its dependence on the inhibited route for its elimination, the greater is the potential for an altered pharmacokinetics of the drug. If more than 70% of a drug is eliminated using the inhibited pathway, the risk for interaction is high (Dunne et al., 2011). Thus, it is important to balance the clearance of a victim drug between hepatic, renal, and biliary routes during the design stage, in order to minimize risk from drug–drug interactions. For a high clearance victim drug that is orally administered, the magnitude of interaction is likely to be high due to first‐pass, making it more sensitive to DDI compared with a low clearance drug. Victim drugs associated with a narrow therapeutic window and high variability are more sensitive to DDI.
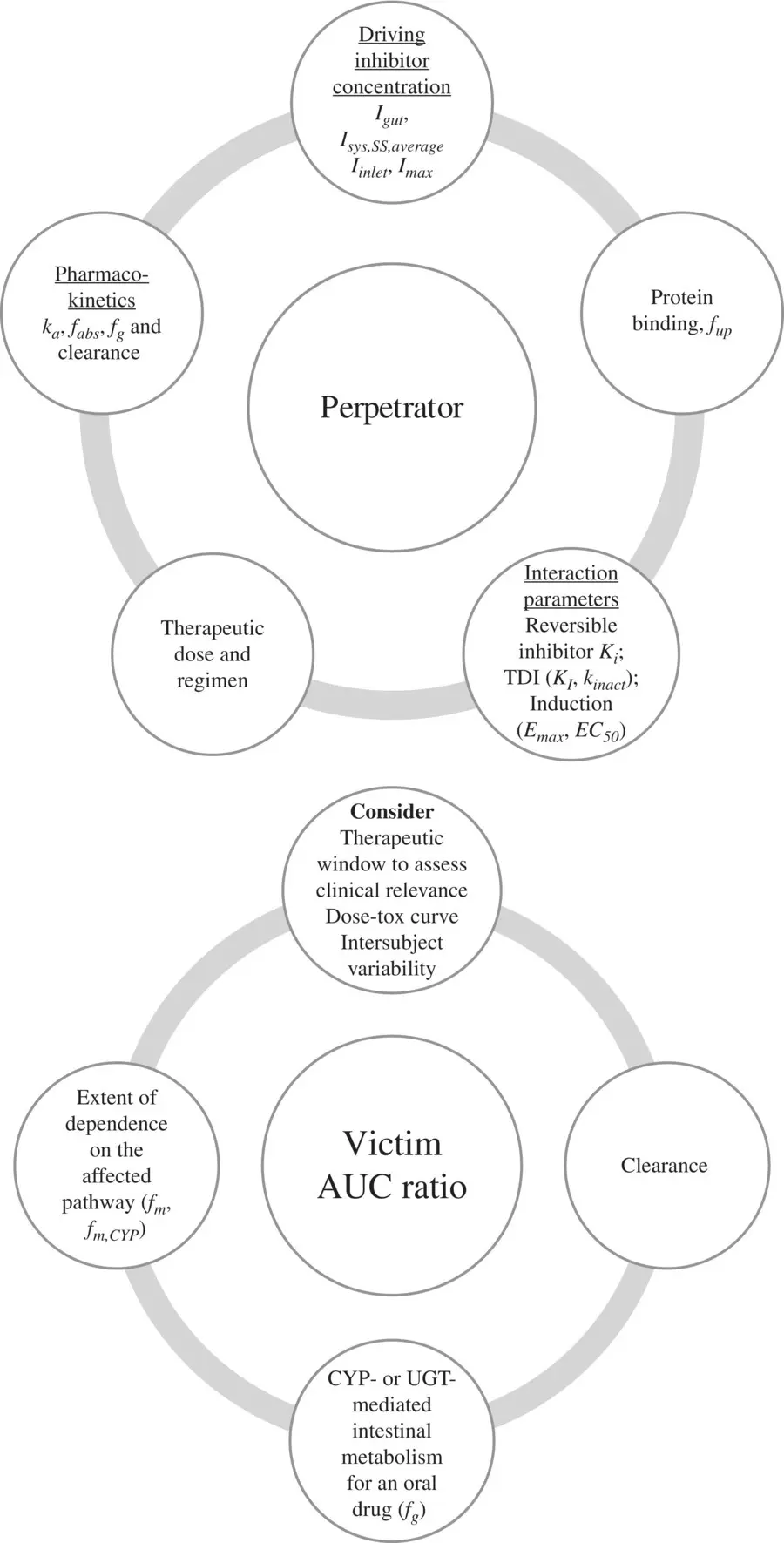
Figure 2.2. Perpetrator and victim properties impacting drug interaction risk.
Induction and/or inhibition of drug transporters of the small intestine, liver, and kidney are major determinants of drug–drug interactions. Transporter‐mediated drug–drug interactions in these three organs can considerably influence the pharmacokinetics and clinical effects of drugs. Transporters of interest are those controlling intestinal efflux, hepatic efflux, blood–brain barrier (P‐gp and BCRP), hepatic uptake (OATP1B1 and OATP1B3) and renal tubular uptake/bidirectional transport (OAT1, OAT3, OCT2), and efflux (MATE1 and MATE2/K). Renal DDIs are rare and associated with significantly lower AUC ratios compared with hepatic DDIs. Victims of renal DDIs are generally compounds whose eliminations are largely dependent on the renal route. Examples include metformin, a substrate of OCT2 and MATEs, and the endogenous compound creatinine, a substrate of OAT2, OCT2, and MATEs, the secretion of both of which are reduced by cimetidine, known to inhibit OCT2 and MATEs. Cimetidine also reduces the renal clearance of bisoprolol, nicotine, and procainamide (Ayrton and Morgan, 2001; Shitara et al., 2009; Shitara et al., 2009; Kirch et al., 1987; Somogyi et al., 1987; Bendayan et al., 1990; Ayrton and Morgan, 2001; Shitara et al., 2009; Ivanyuk et al., 2017). Uptake transporters act in concert with efflux transporters to eliminate toxins (e.g., OCT2 and MATE1, MATE2‐K). Therefore, for secreted drugs that are reliant on transporters for overcoming the membrane barrier, the inhibition of efflux transporters could result in an accumulation of drugs in the cytoplasm of proximal tubular cells, leading to toxic effects in the kidney (nephrotoxicity). Perpetrators of renal DDIs are usually acids or bases with high therapeutic doses and inhibit the renal uptake transporters OATs 1 and 3 or OCT2. Probenecid inhibits the renal OATs and causes drug–drug interactions when coadministered with hydrophilic acids that are predominantly cleared by the kidney, such as penicillin, famotidine, and chlorothiazide (Inotsume et al., 1990; Ayrton and Morgan, 2001; Ho and Kim, 2005; Shitara et al., 2009). Decreases in renal function with age and disease will impact the extent of drug interaction.
Complex drug–drug interactions (DDIs) with potential involvement of multiple elimination pathways and interaction mechanisms is challenging. When a transporter and an enzyme are involved in DDI, then the net effect depends on whether the inhibitor or the substrate of the enzyme is dependent on the transporter for access to the enzyme. Understanding the mechanisms underlying complex DDIs are challenged by the lack of specific probe substrates for transporters (Jones et al., 2020). Rifampicin through its activation of the nuclear receptor, PXR, induces CYP3A4. Additionally, it also inhibits OATP1B1 uptake transporter. Thus, a single dose of rifampicin caused an 8‐fold increase in the in the exposure of atorvastatin (Lau et al., 2007) However, multiple doses of rifampicin caused a decrease in the exposure of atorvastatin, due to induction (Backman et al., 2005). When multiple inhibitors inhibit multiple enzymes, the overall effect is additive, but if they inhibit the same enzyme, then the net effect defaults to the most potent inhibitor.
2.4 IN VITRO METHODS TO EVALUATE DRUG–DRUG INTERACTIONS
A candidate drug must be evaluated for its potential to be a perpetrator or victim of DDI with likely coadministered drugs. In vitro methods that aid this assessment focus on reversible/irreversible inhibition or induction mediated by an enzyme or a transporter.
2.4.1 Candidate Drug as a Potential Perpetrator
Determination of IC 50 and K i for reversible enzyme‐ or transporter‐mediated inhibition:A commonly used measure of inhibitory potential of a drug is the inhibitor concentration required to inhibit 50% of the metabolic rate of a probe substrate ( IC 50) in vitro. IC 50can be converted to absolute inhibition constant ( K i) using the Cheng–Prusoff equation (Yung‐Chi and Prusoff, 1973). For a competitive enzyme inhibition, K iis given as IC 50/(1+[ S / K m]), where S is the substrate concentration and K mis the Michaelis–Menten constant for the substrate. Thus, when measurement of IC 50is done at substrate concentrations that are less than or approaching the K m, IC 50is less than K ifor a reversible enzyme inhibition. When the substrate concentration exceeds its K m, IC 50will overestimate K i. For noncompetitive inhibition, IC 50= K i,and enzyme inhibition is independent of substrate concentration. Assumption of the mechanism of inhibition is thus necessary to get K ifrom IC 50. The in vitro models employed to study enzyme inhibition are hepatocytes or microsomes, depending on the role of phase II enzymes in the metabolism of the substrate. The advantage of these systems is the presence of enzymes in proportions found in vivo. A disadvantage of these systems is that only partial inhibition can be observed for substrates lacking specificity. In these cases, recombinant systems can be useful. Partial inhibition is also attributable to poor solubility of the inhibitor. The preferred in vitro systems for quantifying inhibitory potency of transporter inhibitors are Caco‐2 cells (do not distinguish between transporters), HEK, or MDCK transfected cells for uptake transporters and membrane vesicles for efflux transporters. The inhibitor and substrate concentrations used in the in vitro assays should be clinically relevant. If the inhibitor concentrations in the assay are in excess of the clinical concentrations, the inhibitory potential determined would overestimate the risk in vivo. If the substrate concentration is different from clinically relevant plasma concentrations, then the relative contribution of different enzymes involved in the substrate metabolism could be different from that in vivo, making the in vitro IC 50irrelevant.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations»
Представляем Вашему вниманию похожие книги на «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.