Tina M. Henkin - Snyder and Champness Molecular Genetics of Bacteria
Здесь есть возможность читать онлайн «Tina M. Henkin - Snyder and Champness Molecular Genetics of Bacteria» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Snyder and Champness Molecular Genetics of Bacteria
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Snyder and Champness Molecular Genetics of Bacteria: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Snyder and Champness Molecular Genetics of Bacteria»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Although the text is centered on the most-studied bacteria,
and
, many examples are drawn from other bacteria of experimental, medical, ecological, and biotechnological importance. The book's many useful features include
Text boxes to help students make connections to relevant topics related to other organisms, including humans A summary of main points at the end of each chapter Questions for discussion and independent thought A list of suggested readings for background and further investigation in each chapter Fully illustrated with detailed diagrams and photos in full color A glossary of terms highlighted in the text While intended as an undergraduate or beginning graduate textbook, Molecular Genetics of Bacteria is an invaluable reference for anyone working in the fields of microbiology, genetics, biochemistry, bioengineering, medicine, molecular biology, and biotechnology.
"This is a marvelous textbook that is completely up-to-date and comprehensive, but not overwhelming. The clear prose and excellent figures make it ideal for use in teaching bacterial molecular genetics."—
, University of Washington
Snyder and Champness Molecular Genetics of Bacteria — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Snyder and Champness Molecular Genetics of Bacteria», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
To accommodate the fact that the two DNA polymerases must move in opposite directions and still remain tethered, the lagging-strand template probably loops out as an Okazaki fragment is synthesized. The loop is then relaxed as the polymerase on the lagging-strand template is released from the β clamp, allowing the DNA polymerase to rapidly and efficiently “hop” ahead to the next RNA primer to begin synthesizing the next Okazaki fragment ( Figure 1.10). The polymerase associates with a new β clamp assembled by the clamp loader at the site of the new RNA primer, while the old β clamp is left behind. The β clamp left on the last Okazaki fragment plays important roles in finishing synthesis and joining the fragments of lagging-strand DNA via interactions with DNA polymerase I, ligase, and repair proteins. β clamps are eventually recycled, possibly through the removal function of the δ subunit of the clamp loader. This model involving the looping out of the lagging-strand template has been referred to as the “trombone” model of replication because the loops forming and contracting at the replication fork resemble the extension and return of the slide of the musical instrument. The situation is probably similar in all bacteria and even the other domains of life, although in some other bacteria, including Bacillus subtilis , and in eukaryotes, different combinations of DNA polymerases are used to polymerize the leading and lagging strands (see Sanders et al., Suggested Reading). In the case of B. subtilis , an additional DNA polymerase interacts with DnaG and the replicative helicase, extending the RNA primer with DNA before handing the template off to the DNA polymerase used for the majority of DNA replication on both strands.
In addition to its role in loading β clamps onto template DNA, the clamp loader also plays an important role in coordinating the various replication components. Not only does the τ subunit of the clamp loader interact with DNA polymerase on the leading-strand and laggingstrand templates, it also interacts with the DnaB helicase ( Figure 1.10). Further coordination on the lagging-strand template is facilitated by interactions between the DnaG primase and DnaB helicase ( Figure 1.10). Coordination through the clamp loader helps to focus the energy from DNA polymerization with the energy that powers the helicase, allowing a high rate of DNA replication. The interaction between DNA polymerase III and DnaB governs the speed of unwinding so that it matches the rate of DNA polymerization to prevent undue exposure of singlestranded DNA.
THE GENES FOR REPLICATION PROTEINS
Most of the genes for replication proteins have been found by isolating mutants defective in DNA replication, but not RNA or protein synthesis. Since a mutant cell that cannot replicate its DNA will die, any mutation (for definitions of mutants and mutations, see “Replication Errors” below and chapter 3) that inactivates a gene whose product is required for DNA replication will kill the cell. Therefore, for experimental purposes, only a type of mutant called a temperature-sensitive mutantcan be usefully isolated with mutations in DNA replication genes. These are mutants in which the product of the gene is active at one temperature but inactive at another. The mutant cells can be propagated at the temperature at which the protein is active (the permissive temperature). However, shifting to the other (nonpermissive) temperature can test the effects of inactivating the protein. The molecular basis of temperature-sensitive mutants is discussed in more detail in chapter 3.
The immediate effect of a temperature shift on a mutant with a mutation in a DNA replication gene depends on whether the product of the gene is continuously required for replication at the replication forks or is involved only in the initiation of new rounds of replication. For example, if the mutation is in a gene for DNA polymerase III or in the gene for the DnaG primase, replication ceases immediately. However, if the temperature-sensitive mutation is in a gene whose product is required only for initiation of DNA replication, for example, the gene for DnaA or DnaC (see “Initiation of Chromosome Replication” below), the replication rate for the population will slowly decline. Unless the cells have been somehow synchronized in their cell cycles, each cell is at a different stage of replication, with some cells having just finished a round of replication and other cells having just begun a new round. Cells in which rounds of chromosome replication were under way at the time of the temperature shift will complete their replication cycle but will not start a new round. Therefore, the rate of replication decreases until the rounds of replication in all the cells are completed.
Replication Errors
To maintain the stability of a species, replication of the DNA must be almost free of error. Changes in the DNA sequence that are passed on to subsequent generations are called mutations(see chapter 3). Depending on where these changes occur, they can severely alter the protein products of genes or other cellular functions. To avoid such instability, the cell has mechanisms that reduce the error rate.
As DNA replicates, the wrong base is sometimes inserted into the growing DNA chain. For example, Figure 1.11shows the incorrect incorporation of a T opposite a G. Such a base pair in which the bases are paired wrongly is called a mismatch. Mismatches can occur when the bases take on forms called tautomers, which pair differently from the normal form of the base (see chapter 3). After the first replication shown in Figure 1.11, the mispaired T pairs with an A, causing a GC-to-AT change in the sequence of one of the two progeny DNAs and thus changing the base pair at that position on all subsequent copies of the mutated DNA molecule.
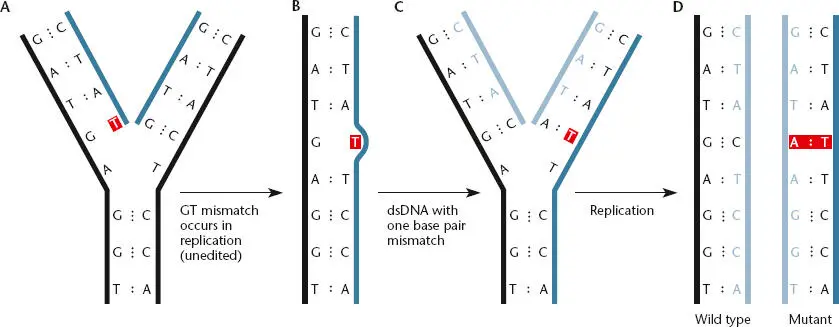
Figure 1.11 Mistakes in base pairing can lead to changes in the DNA sequence called mutations. If a T is mistakenly placed opposite a G during replication (A), it can lead to an AT base pair replacing a GC base pair in the progeny DNA (B to D).
Editing
One way the cell reduces mistakes during replication is through editing functions. With some DNA polymerases the editing function resides in the same protein, while in other cases a separate protein performs the editing function. Editing proteins are aptly named because they go back over the newly replicated DNA looking for mistakes and then recognize and remove incorrectly inserted bases ( Figure 1.12). If the last nucleotide inserted in the growing DNA chain creates a mismatch, the editing function stops replication until the offending nucleotide is removed. DNA replication then continues, inserting the correct nucleotide. Because the DNA chain grows in the 5′-to-3′ direction, the last nucleotide added is at the 3′ end. The enzyme activity found in a DNA polymerase or one of its accessory proteins that removes this nucleotide is therefore called a 3′ exonuclease. The editing proteins probably recognize a mismatch because the mispairing (between T and G in the example in Figure 1.11) causes a minor distortion in the structure of the double-stranded helix of the DNA.
DNA polymerase I is an example of a DNA polymerase in which the 3′ exonuclease editing activity is part of the DNA polymerase itself. However, in DNA polymerase III, which replicates the bacterial chromosome, the editing function resides in an accessory protein encoded by a separate gene whose product travels along the DNA with the DNA polymerase during replication. In E. coli , the 3 ' exonuclease editing function is encoded by the dnaQ gene (Table 1.1), and dnaQ mutants, also called mutD mutants (i.e., cells with a mutation in this gene that inactivates the 3 ' exonuclease function), show much higher rates of spontaneous mutagenesis than do cells containing the wild-type, or normally functioning, dnaQ gene product. Because of their high spontaneous mutation rates, mutD mutants of E. coli can be used as a tool for mutagenesis, often combined with mutations in other genes whose products normally contribute to the correction of mismatches (see chapter 10).
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Snyder and Champness Molecular Genetics of Bacteria»
Представляем Вашему вниманию похожие книги на «Snyder and Champness Molecular Genetics of Bacteria» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Snyder and Champness Molecular Genetics of Bacteria» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.