Jane Flint - Principles of Virology
Здесь есть возможность читать онлайн «Jane Flint - Principles of Virology» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.
- Название:Principles of Virology
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:3 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Principles of Virology: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Principles of Virology»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Volume I: Molecular Biology
Volume II: Pathogenesis and Control
Principles of Virology, Fifth Edition
Principles of Virology — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Principles of Virology», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Other genome-wide mapping analyses that can be performed include identifying the binding sites for long noncoding RNAs (lncRNA) on chromatin using capture hybridization analysis of RNA targets (CHART). In this method, biotin-linked oligonucleotides that are complementary to the target RNA are designed. These are added to reversibly cross-linked chromatin extracts, and the target RNA is purified with streptavidin beads, which bind with high afnity to biotin. The sequences of the RNA targets identify the genomic binding sites of endogenous RNAs. A related method is chromatin isolation by RNA purification (ChIRP), in which tiled oligonucleotides labeled with biotin are used to retrieve specific lncRNA bound to protein and DNAs.
How DNA is organized in virus particles and in the cell nucleus is being studied using chromosome conformation capturetechnology, abbreviated as 3C, 4C, 5C, and Hi-C, which differ in scope. For example, 3C identifies interactions between a single pair of genomic loci. Chromosome conformation capture on chip (4C) studies the interaction of one genomic locus and all other genomic loci, while chromosome conformation capture carbon copy (5C) detects interactions between all restriction fragments in a given region. In HiC, high-throughput sequencing is used to identify the restriction fragments studied. These methods begin with cross-linking of cell genomes with formaldehyde and digestion with restriction endonucleases, followed by random ligation under conditions where joining of cross-linked fragments is favored over those that are not. PCR is then used to amplify ligated junctions and identify interacting loci. The open or closed state of chromatin can be measured by DNaseI-seq(DNaseI hypersensitive sites sequencing) and FAIRE-seq(formaldehyde-assisted isolation of regulatory elements). These protocols are based on the use of formaldehyde to cross-link DNA: this reaction is more efficient in nucleosome-rich regions than in nucleosome-poor areas. The non-cross-linked DNA, typically from open chromatin, is then purified and its sequence is determined. The two protocols differ in that FAIRE-seq does not require permeabilization of cells or the isolation of nuclei. The methylation state of DNA can be assessed using bisulfite sequencing. Treatment of DNA with bisulfite converts C to U but does not affect 5-methylated cytosines. A variety of sequencing methods that can use this change to provide single-nucleotide resolution information about DNA methylation have been developed. As might be expected, interpreting the growing sets of data on chromatin structure has required the development of new statistical and computational approaches.
Mass Spectrometry
Mass spectrometry (MS) is a technique that can identify the chemical constituents of complex and simple mixtures. It has emerged as a powerful tool for detecting and quantifying thousands of proteins in biological samples, including viruses and virus-infected cells.
A mass spectrometer ionizes the chemical constituents of a mixture and then sorts the ions based on their mass-to-charge ratio. Identification of the components is done by comparison with the patterns generated by known materials.
The total protein content of a cell or a virus particle is called the proteome. Human cells have been estimated to contain from 500,000 to 3,000,000 proteins per cubic micrometer, encoded by ∼20,000 open reading frames, and their products are further diversified by transcriptional, posttranscriptional, translational, and posttranslational regulation. The cell proteome may be further altered during virus infection. The proteome of virus particles is far less complex, but the very largest viruses can still contain hundreds of proteins. Mass spectrometry can be used to identify proteins and their concentrations in cells and in virus particles and also to reveal protein localization, protein-protein interactions, and posttranslational modifications in infected and uninfected cells.
Mass spectrometry may be combined with biochemical and genomic techniques to provide global views of viral reproduction cycles. For example, changes in proteins secreted by host cells upon virus infection can be readily characterized by performing mass spectrometry on supernatants from infected cells. Another application is to identify protein-protein interactions in virus-infected cells: a promiscuous biotinylating enzyme can be directed to a subcellular compartment, where it biotinylates adjacent molecules. These can be purified by attachment to streptavidin-containing beads and identified by mass spectrometry. Integration of mass spectrometry with some of the methods described above for genome analysis can be used to identify proteins that participate in the regulation of gene expression.
At one time the mass spectrometer was a very expensive instrument restricted to chemistry laboratories. Recent advances in the instrumentation, including cost reduction, as well as sample preparation and computational biology have propelled this technology into the virology research laboratory.
Protein-Protein Interactions
A major goal of virology research is to understand how protein-protein interactions modulate reproduction cycles and pathogenesis. Consequently, multiple experimental approaches have been devised to identify the entire set of interactions among viral proteins and between viral and cell proteins. The yeast two-hybrid screen, a complementation assay which was designed to discover protein-protein interactions, has been adapted to high-throughput applications. In this assay, a transcriptional regulatory protein is split into two fragments, the DNA-binding domain and the activating domain. The coding sequences of two different proteins are fused with the two domains. If the two proteins interact, when the fusion proteins are produced in cells, transcriptional activation (leading to the transcription of a reporter gene) will take place. For high-throughput applications, libraries of protein-coding DNAs are screened against a single viral protein or all viral proteins. This method was used to describe the virus-host interactome of two herpesviruses.
Other approaches to defining interactomes include coimmunoprecipitation, affinity purification of tagged proteins ( Fig. 2.21), and labeling of cell proteins with chemical cross-linkers (used to identify plant proteins that interact with plant virus proteins), followed by mass spectrometry.
While these methods allow definition of virus-cell interactomes, they are not unambiguous. For at least one virus, interactomes determined in different laboratories are very diverse. Most importantly, the observation of a protein-protein interaction does not confirm biological relevance: the roles of such interactions in viral reproduction must be determined by other means ( Box 2.12).
Single-Cell Virology
Much of virology research is carried out by using populations of cells in culture or in animals. However, as discovered by virologists in the 1950s, individual cells of the same type can behave very differently with respect to susceptibility and permissiveness to infection and the kinetics of virus production.
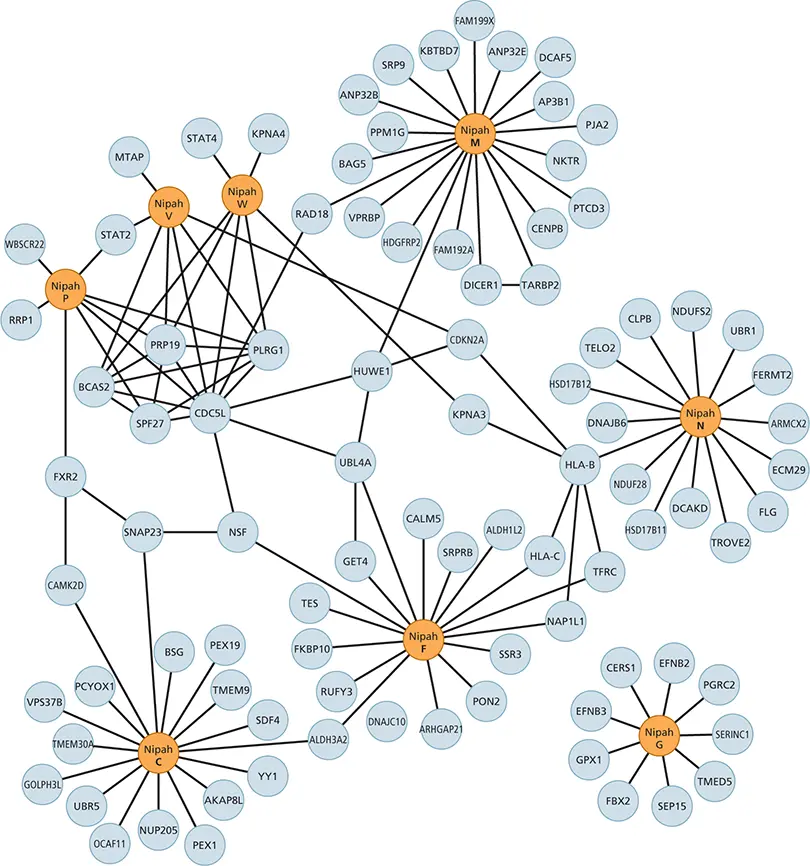
Figure 2.21 Interactions between human proteins and Nipah virus proteins. Network representation of interactions of Nipah virus and human proteins determined by affinity purification and mass spectrometry. Nipah virus proteins are shown in orange. Cellular proteins are shown in gray. Protein names (from UniProt) are shown. Adapted from Martinez-Gil L, Vera-Velasco NM, Mingarro I. 2017. J Virol 91:e01461-17, with permission.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Principles of Virology»
Представляем Вашему вниманию похожие книги на «Principles of Virology» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Principles of Virology» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.