Handbook of Aggregation-Induced Emission, Volume 3
Здесь есть возможность читать онлайн «Handbook of Aggregation-Induced Emission, Volume 3» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Handbook of Aggregation-Induced Emission, Volume 3
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Handbook of Aggregation-Induced Emission, Volume 3: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Handbook of Aggregation-Induced Emission, Volume 3»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
the editors address the applications of AIEgens in several fields, including bio-imaging, fluorescent molecular switches, electrochromic materials, regenerative medicine, detection of organic volatile contaminants, hydrogels, and organogels. Topics covered include:
AIE-active emitters and their applications in OLEDs, and circularly polarized luminescence of aggregation-induced emission materials AIE polymer films for optical sensing and energy harvesting, aggregation-induced electrochemiluminescence, and mechanoluminescence materials with aggregation-induced emission Dynamic super-resolution fluorescence imaging based on photoswitchable fluorescent spiropyran Visualization of polymer microstructures Self-assembly of micelle and vesicles New strategies for biosensing and cell imaging Perfect for academic researchers working on aggregation-induced emission, this set of volumes is also ideal for professionals and students in the fields of photophysics, photochemistry, materials science, optoelectronic materials, synthetic organic chemistry, macromolecular chemistry, polymer science, and biological sciences.
Handbook of Aggregation-Induced Emission, Volume 3 — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Handbook of Aggregation-Induced Emission, Volume 3», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Another important example of oxidative‐reductive ECL is based on the use of alkylamines. [Ru(bpy) 3] 2+or its derivatives with tri‐ n ‐propylamine (TPrA) as coreactant, exhibits the highest ECL efficiency and represents the most common luminophore/coreactant couple, which forms the basis of commercial immunoassays and DNA analyses [3, 4], and it can be considered as an ECL standard. Noffsinger and Danielson have first reported the [Ru(bpy) 3] 2+ECL reaction with alkylamines [41], and in 1990 Leland and Powell first reported the use of TPrA with Ru(bpy) 3 2+to produce highly intense ECL [12]. The experiment was carried out on a gold electrode in a buffer solution of TPrA and Ru(bpy) 3 2+.
Also, to generate AI‐ECL the employment of trialkylamines was chosen many times, and examples will be clarified in Section 4.3[42–44].) Coreactant ECL using trialkylamines can proceed through several possible parallel routes. One pathway for AS‐TPrA coreactant ECL is represented by the following reactions [12, 45, 46]:
(4.10) 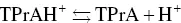
(4.11) 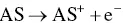
(4.12) 
(4.13) 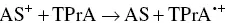
(4.14) 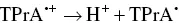
(4.15) 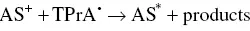
followed by Equation 4.9.
When the concentration of TPrA is high and the concentration of AS is low, as is generally the case when the luminophore is being used as a probe in a bioanalytical context, an alternative pathway emerges [29].
(4.16) 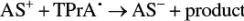
(4.17) 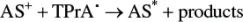
Here, TPrA •generates the reduced form of the luminophore via homogeneous electron transfer ( Equation 4.11), following which, the excited state may be formed via reaction, according to Equation 4.3(annihilation), or by reaction with the radical cation TPrA •+( Equation 4.17).
In general, there is a strong dependence of ECL intensities on pH according to analytes. Oxalates show virtual independence of ECL intensities on sample pH but are mostly detected at pH 6.0 [47]. Alkylamines produce maximum intensities between pH 4.5 and 6.0 [48, 49].
The reductive‐oxidative mostly employed mechanism concerning K 2S 2O 8, which has been firstly explored by White and Bard in 1982 [50], and used in AI‐ECL of siloles [51]. K 2S 2O 8offers some advantages with respect to amines. Because S 2O 8 2−does not react appreciably with water or oxygen, it appears particularly promising for aqueous ECL, where the decomposition of water generally prohibits the direct production by electrolysis of the reactant that generates the excited state. The pathways are summarized by the Equations 4.18– 4.21:
(4.18) 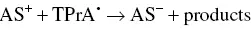
(4.19) 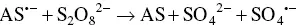
(4.20) 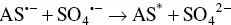
(4.21) 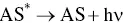
At first, AS gets reduced to generate the radical anion AS •−( Equation 4.18). Then, the reaction between this radical anion and S 2O 8 2−produces the strong oxidant species SO 4 •−( Equation 4.19). Following the electron transfer between the AS •−and SO 4 •−that generates the excited state ( Equation 4.20) that finally emits light ( Equation 4.21).
4.2 Classification and Properties of AI‐ECL luminophores
From our discovery in 2017 [28], a huge attention has been paid to the development of different AI‐ECL luminophores, considering the background knowledge in both ECL and AIE. Since the aggregation form can be obtained in aqueous systems, ECL properties have been mostly evaluated by following the coreactant mechanism. Up to today, the reported samples based on inorganic materials or polymeric aggregated forms belong to the oxidative‐reductive pathway, while few of the organic materials show reductive‐oxidative pathway. Following, AI‐ECL luminophores are classified by typology of material or molecule. Metal transition complexes include platinum and iridium complexes, polymers and polymeric nanoaggregates can be entirely organic or can include metal transition complex moieties. The polymer has the advantage of easy‐forming dots or nanoparticles by reprecipitation a microemulsion. The same fate is observed for organic molecules while different hybrid materials have been synthesized in other ways to obtain them with various sizes and shapes.
4.2.1 Metal Transition Complexes
The first investigation of AI‐ECL was reported by our group on square‐planar platinum(II) complexes and their supramolecular assemblies in solution and solid‐state ( Figure 4.4) [28]. The two complexes presented, 1and 2in Figures 4.4a–d, maintain the same tridentate ligand 2,6‐bis(3‐[trifluoromethyl]‐1 H ‐1,2,4‐triazol‐5‐yl)pyridine (pyC 5‐CF 3‐tzH 2), and differ on the number of triethylene glycols chain attached to a 4‐amino pyridine ancillary ligand. It was demonstrated from the same group that this class of complexes exhibits very poor structured blue emission in a nonpolar solvent like dichloromethane with photoluminescence quantum yield (φ PL) around 1% and short lifetimes [52, 53]. When they were dissolved in aqueous media, photoluminescent properties showed a significant bathochromical shift to 600 nm and an enhancement of both φ PLand lifetime, up to 70–80%. The group attributed these results to the formation of aggregates in water, which approached single metal complexes from a distance shorter than 3.5 Å. The Pt⋯Pt metallophilic interactions at that distance caused the destabilization of the d z 2orbitals and the population of a new metal‐based highest occupied molecular orbital (HOMO) leading to metal‐metal‐to‐ligand charge transfer (MMLCT) transitions. While 1formed spherical aggregates not thermodynamically stable in water, complex 2displayed a stable aqueous suspension of nanoaggregates of 20 nm, which were relatively small to diffuse at the electrode surface and got oxidized. As we said, the Pt⋯Pt interaction destabilizes the HOMO and from an electrochemical point of view it decreases the oxidation potential of complex 2, which was not falling in the electrochemical window of dichloromethane but it is visible at +1.33 V vs Ag/AgCl in water ( Figure 4.4e). In fact, upon oxidation and in presence of a coreactant, these nanoaggregates resulted in extremely active ECL compared to the molecularly dissolved complex in dichloromethane or acetonitrile with an ECL efficiency compared to Ru(bpy) 3 2+of Equation 4.2using TPrA as coreactant and Equation 1.14 with C 2O 4 2−. Despite the impossibility to use complex 1in solution, it was possible to obtain the same kind of Pt⋯Pt interaction in solid‐state. It is in fact well known that these complexes manifest similar mechanochromical properties [54, 55]. Therefore, it was demonstrated that AI‐ECL emission could be obtained from complex 1onto screen‐printed electrodes (SPEs) after its blue‐emitting powder was mechanical pressed to generate the desired interaction and therefore an orange solid photoluminescence (PL). The same light could be collected upon electrochemical oxidation in presence of C 2O 4 2−obtaining an enhancement of a factor 20 from the blue powder ( Figure 4.4c). It is worthful to say that C 2O 4 2−acts as a better coreactant because it stabilizes the platinum complex when Pt 2+is oxidized to Pt 4+changing the geometry from square planar to tetrahedral [56]. The change in coordination geometry could be a limitation in the use of these complex‐types for multiple scans and further studies are required to understand deeper mechanism behind the generation of the MMLCT excited state by electrochemistry, since a structural change is happening during the process.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Handbook of Aggregation-Induced Emission, Volume 3»
Представляем Вашему вниманию похожие книги на «Handbook of Aggregation-Induced Emission, Volume 3» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Handbook of Aggregation-Induced Emission, Volume 3» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.