Michael J. Neal - Medical Pharmacology at a Glance
Здесь есть возможность читать онлайн «Michael J. Neal - Medical Pharmacology at a Glance» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Medical Pharmacology at a Glance
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Medical Pharmacology at a Glance: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Medical Pharmacology at a Glance»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
is the ideal companion for all medical and healthcare students, providing a visual overview of pharmacology, and describing the basic principles of drug action, interaction, absorption, and excretion. Clear and accessible chapters organised around common diseases and conditions facilitate efficient clinical learning, and include references to drug classes and side effects, disease pathophysiology, prescribing guidelines, and more.
Now in its ninth edition, this leading guide has been thoroughly updated to reflect current guidelines and drug information. This edition features new and revised illustrations, additional pedagogical tools, and enhanced online content. Widely recognised as both the best introduction to medical pharmacology and the perfect revision tool for USMLE and pharmacology exams, this invaluable guide:
Covers a wide range of drugs used to treat conditions such as hypertension, anaemias, cancer, and affective disorders Explains drug mechanisms and the principles of drug action Discusses practical topics including drug misuse, drug indications, and side effects Includes a companion website featuring online cases, flashcards, and a list of core drugs
Medical Pharmacology at a Glance — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Medical Pharmacology at a Glance», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
The ionization of weak acids and bases depends on the pH of the tubular fluid. Manipulation of the urine pH is sometimes useful in increasing renal excretion. For example, bicarbonate administration makes the urine alkaline; this ionizes aspirin, making it less lipid soluble and increasing its rate of excretion.
Weak acids and weak bases are actively secreted in the proximal tubule, e.g. penicillins, thiazide diuretics, morphine.
Biliary excretionSome drugs (e.g. diethylstilbestrol) are concentrated in the bile and excreted into the intestine where they may be reabsorbed. This enterohepatic circulation increases the persistence of a drug in the body.
4 Drug metabolism
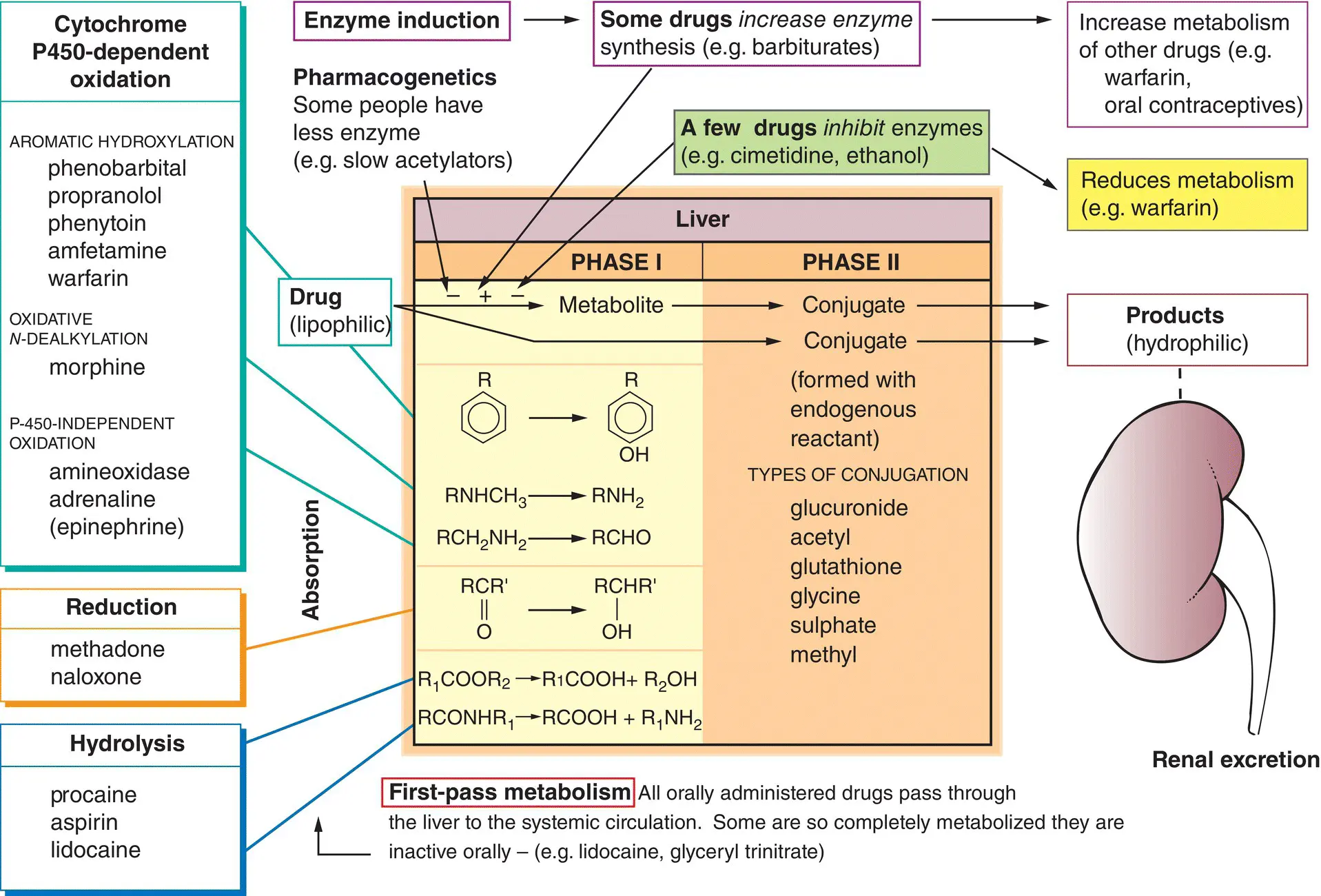
Drug metabolism has two important effects.
1 The drug is made more hydrophilic – this hastens its excretion by the kidneys (right, ) because the less lipid‐soluble metabolite is not readily reabsorbed in the renal tubules.
2 The metabolites are usually less active than the parent drug. However, this is not always so, and sometimes the metabolites are as active as (or more active than) the original drug. For example, diazepam (a drug used to treat anxiety) is metabolized to nordiazepam and oxazepam, both of which are active. Prodrugs are inactive until they are metabolized in the body to the active drug. For example, levodopa, an antiparkinsonian drug ( Chapter 26), is metabolized to dopamine, whereas the hypotensive drug methyldopa ( Chapter 15) is metabolized to α‐methylnorepinephrine.
The liveris the main organ of drug metabolism and is involved in two general types of reaction.
Phase I reactions
These involve the biotransformation of a drug to a more polar metabolite (left of the figure) by introducing or unmasking a functional group (e.g. –OH, –NH 2, –SH).
Oxidationsare the most common reactions and these are catalysed by an important class of enzymes called the mixed function oxidases ( cytochrome P450s). The substrate specificity of this enzyme complex is very low and many different drugs can be oxidized (examples, top left). Other phase I reactions are reductions(middle left) and hydrolysis(bottom left).
Phase II reactions
Drugs or phase I metabolites that are not sufficiently polar to be excreted rapidly by the kidneys are made more hydrophilic by conjugation with endogenous compounds in the liver (centre of the figure).
Repeated administration of some drugs (top) increases the synthesis of cytochrome P450 ( enzyme induction). This increases the rate of metabolism of the inducing drug and also of other drugs metabolized by the same enzyme (top right). In contrast, drugs sometimes inhibitmicrosomal enzyme activity (top,  ) and this increases the action of drugs metabolized by the same enzyme (top right,
) and this increases the action of drugs metabolized by the same enzyme (top right,  ).
).
In addition to these drug–drug interactions, the metabolism of drugs may be influenced by genetic factors(pharmacogenetics), age and some diseases, especially those affecting the liver.
Drugs
Most drugs are highly lipophilic and are often bound to plasma proteins. As the protein‐bound drug is not filtered at the renal glomerulus and the free drug readily diffuses back from the tubule into the blood, such drugs would have a very prolonged action if their removal relied on renal excretion alone. In general, drugs are metabolized to more polar compounds, which are more easily excreted by the kidneys. A few drugs are highly polar because they are fully ionized at physiological pH values. Such drugs are metabolized little, if at all, and the termination of their actions depends mainly on renal excretion.
Liver
The main organ of drug metabolism is the liver, but other organs, such as the gastrointestinal tract and lungs, have considerable activity. Drugs given orally are usually absorbed in the small intestine and enter the portal system to travel to the liver, where they may be extensively metabolized (e.g. lidocaine, morphine, propranolol). This is called first‐pass metabolism , a term that does not refer only to hepatic metabolism. For example, chlorpromazine is metabolized more in the intestine than by the liver.
Phase I reactions
The most common reaction is oxidation . Other, relatively uncommon, reactions are reduction and hydrolysis .
The P450 monooxygenase system
Cytochrome P450 enzymes form a superfamily of related enzymes that differ in amino acid sequence. Each is referred to as CYP followed by defining numbers and a letter. There are over 70 CYP gene families but only three are involved in hepatic drug metabolism (CYP1, CYP2 and CYP3). Oxidation by the P450 monooxygenase system is complex but the result is simple, the addition of an –OH group to the drug. Numerous (CYP) isoforms of P450 exist with different, but often overlapping, substrate specificities. About half a dozen P450 isoforms account for most hepatic drug metabolism. CYP3A4 is worth remembering because it metabolizes more than 50% of drugs.
Phase II reactions
These usually occur in the liver and involve conjugation of a drug or its phase I metabolite with an endogenous substance. The resulting conjugates are almost always less active and are polar molecules that are readily excreted by the kidneys.
Factors affecting drug metabolism
Enzyme induction
The activity of some drugs, for example, oestrogen and progesterone may be significantly reduced by a second drug that increases the activity of drug‐metabolizing enzymes (primarily CYP2C9, CYP2C19 and CYP3A4). Phenytoin , carbamazepine and rifampicin are the most potent enzyme inducers. The mechanism involved is unclear but is similar to hormones that bind to response elements in DNA and promote transcription of the appropriate gene. However, not all enzymes subject to induction are microsomal. For example, hepatic alcohol dehydrogenase occurs in the cytoplasm.
Enzyme inhibition
Enzyme inhibition may cause adverse drug interactions. These interactions tend to occur more rapidly than those involving enzyme induction because they occur as soon as the inhibiting drug reaches a high‐enough concentration to compete with the affected drug. Drugs may inhibit different forms of cytochrome P450 and so affect the metabolism only of drugs metabolized by that particular isoenzyme. Cimetidine inhibits the metabolism of several potentially toxic drugs including phenytoin, warfarin and theophylline. Erythromycin also inhibits the cytochrome P450 system and increases the activity of theophylline, warfarin, carbamazepine and digoxin. Substances in the diet can also affect the metabolism of drugs. For example, a component of grapefruit juice inhibits CYP3A4 and may increase the effects of several drugs, such as midazolam, simvastatin.
Genetic polymorphisms
The study of how genetic determinants affect drug action is called pharmacogenetics . The response to drugs may vary significantly between individuals. For example, about 8% of the population has faulty expression of CYP2D6, the P450 isoform responsible for debrisoquine hydroxylation. These poor hydroxylators show exaggerated and prolonged responses to drugs such as propranolol and metoprolol ( Chapter 15), which undergo extensive hepatic metabolism.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Medical Pharmacology at a Glance»
Представляем Вашему вниманию похожие книги на «Medical Pharmacology at a Glance» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Medical Pharmacology at a Glance» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.