Michael J. Neal - Medical Pharmacology at a Glance
Здесь есть возможность читать онлайн «Michael J. Neal - Medical Pharmacology at a Glance» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Medical Pharmacology at a Glance
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Medical Pharmacology at a Glance: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Medical Pharmacology at a Glance»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
is the ideal companion for all medical and healthcare students, providing a visual overview of pharmacology, and describing the basic principles of drug action, interaction, absorption, and excretion. Clear and accessible chapters organised around common diseases and conditions facilitate efficient clinical learning, and include references to drug classes and side effects, disease pathophysiology, prescribing guidelines, and more.
Now in its ninth edition, this leading guide has been thoroughly updated to reflect current guidelines and drug information. This edition features new and revised illustrations, additional pedagogical tools, and enhanced online content. Widely recognised as both the best introduction to medical pharmacology and the perfect revision tool for USMLE and pharmacology exams, this invaluable guide:
Covers a wide range of drugs used to treat conditions such as hypertension, anaemias, cancer, and affective disorders Explains drug mechanisms and the principles of drug action Discusses practical topics including drug misuse, drug indications, and side effects Includes a companion website featuring online cases, flashcards, and a list of core drugs
Medical Pharmacology at a Glance — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Medical Pharmacology at a Glance», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Hormonesare chemicals released into the bloodstream; they produce their physiological effects on tissues possessing the necessary specific hormone receptors. Drugs may interact with the endocrine system by inhibiting (e.g. antithyroid drugs, Chapter 35) or increasing (e.g. oral antidiabetic agents, Chapter 36) hormone release. Other drugs interact with hormone receptors, which may be activated (e.g. steroidal anti‐inflammatory drugs, Chapter 33) or blocked (e.g. oestrogen antagonists, Chapter 34). Local hormones (autacoids), such as histamine, serotonin (5‐hydroxytryptamine, 5HT), kinins and prostaglandins, are released in pathological processes. The effects of histamine can sometimes be blocked with antihistamines ( Chapter 11), and drugs that block prostaglandin synthesis (e.g. aspirin) are widely used as anti‐inflammatory agents ( Chapter 32).
Transport systems
The lipid cell membrane provides a barrier against the transport of hydrophilic molecules into or out of the cell.
Ion channelsare selective pores in the membrane that allow the ready transfer of ions down their electrochemical gradient. The open–closed state of these channels is controlled either by the membrane potential (voltage‐gated channels) or by transmitter substances (ligand‐gated channels). Some channels (e.g. Ca 2+channels in the heart) are both voltage and transmitter gated. Voltage‐gated channels for sodium, potassium and calcium have the same basic structure ( Chapter 5), and subtypes exist for each different channel. Important examples of drugs that act on voltage‐gated channels are calcium‐channel blockers ( Chapter 16), which block L‐type calcium channels in vascular smooth muscle and the heart, and local anaesthetics ( Chapter 5), which block sodium channels in nerves. Some anticonvulsants ( Chapter 25) and some antiarrhythmic drugs ( Chapter 17) also block Na +channels. No clinically useful drug acts primarily on voltage‐gated K +channels, but oral antidiabetic drugs act on a different type of K +channel that is regulated by intracellular adenosine triphosphate (ATP, Chapter 36).
Active transport processesare used to transfer substances against their concentration gradients. They utilize special carrier molecules in the membrane and require metabolic energy. Two examples are listed below.
1 Sodium pump. This expels Na+ ions from inside the cell by a mechanism that derives energy from ATP and involves the enzyme adenosine triphosphatase (ATPase). The carrier is linked to the transfer of K+ ions into the cell. The cardiac glycosides ( Chapter 18) act by inhibiting the Na+/K+‐ATPase. Na+ and/or Cl− transport processes in the kidney are inhibited by some diuretics ( Chapter 14).
2 Norepinephrine transport. The tricyclic antidepressants ( Chapter 28) prolong the action of norepinephrine by blocking its reuptake into central nerve terminals.
Enzymes
These are catalytic proteins that increase the rate of chemical reactions in the body. Drugs that act by inhibiting enzymes include: anticholinesterases , which enhance the action of acetylcholine ( Chapters 6and 8); carbonic anhydrase inhibitors , which are diuretics (i.e. increase urine flow, Chapter 14); monoamine oxidase inhibitors , which are antidepressants ( Chapter 28); and inhibitors of cyclo‐oxygenase (e.g. aspirin, Chapter 32).
Second messengers
These are chemicals whose intracellular concentration increases or, more rarely, decreases in response to receptor activation by agonists, and which trigger processes that eventually result in a cellular response. The most studied second messengers are: Ca 2+ions, cyclic adenosine monophosphate (cAMP), inositol‐1,4,5‐trisphosphate (InsP 3) and diacylglycerol (DAG).
cAMP is formed from ATP by the enzyme adenylyl cyclase when, for example, β‐adrenoceptors are stimulated. The cAMP activates an enzyme (protein kinase A), which phosphorylates a protein (enzyme or ion channel) and leads to a physiological effect.
InsP 3and DG are formed from membrane phosphatidylinositol 4,5‐bisphosphate by activation of a phospholipase C. Both messengers can, like cAMP, activate kinases, but InsP 3does this indirectly by mobilizing intracellular calcium stores. Some muscarinic effects of acetylcholine and α 1‐adrenergic effects involve this mechanism ( Chapter 7).
G‐proteins
G‐protein‐coupled receptors are linked to their responses by a family of regulatory guanosine triphosphate (GTP)‐binding proteins (G‐proteins). The receptor–agonist complex induces a conformational change in the G‐protein, causing its α‐subunit to bind GTP. The α–GTP complex dissociates from the G‐protein and activates (or inhibits) the membrane enzyme or channel. The signal to the enzyme or channel ends because α–GTP has intrinsic GTPase activity and turns itself off by hydrolysing the GTP to guanosine diphosphate (GDP). α–GDP then reassociates with the βγ G‐protein subunits.
2 Drug–receptor interactions
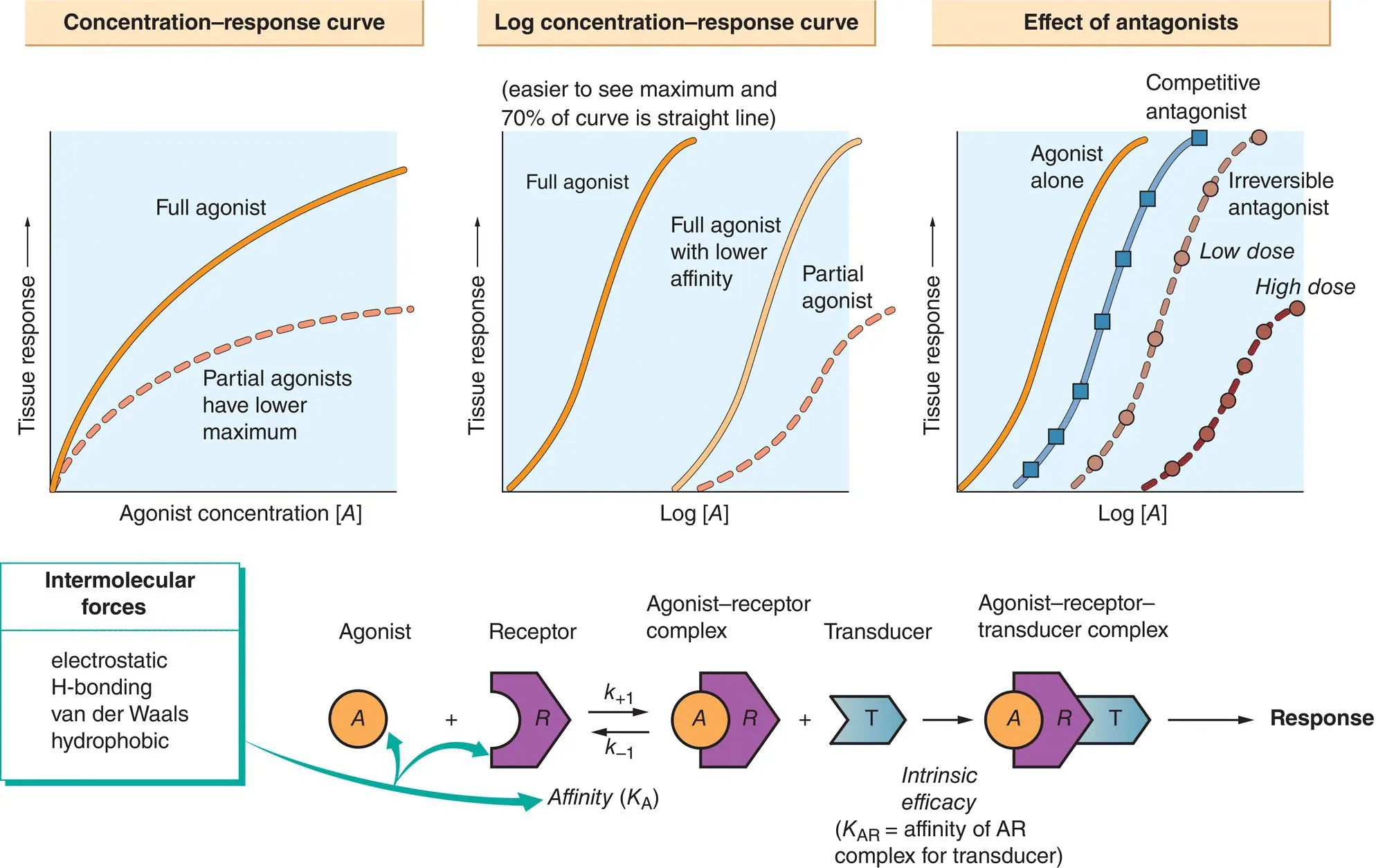
The tissues in the body have only a few basic responses when exposed to agonists (e.g. muscle contraction, glandular secretion), and the quantitative relationship between these physiological responses and the concentration of the agonist can be measured by using bioassays. The first part of the drug–receptor interaction, i.e. the binding of drug to receptor, can be studied in isolation using binding assays.
It has been found by experiment that, for many tissues and agonists, when the response is plotted against the concentration of the drug, a curve is produced that is often hyperbolic ( concentration–response curve, top left). In practice, it is often more convenient to plot the response against the logarithm of the agonist concentration ( log concentration–response curve, middle top). Assuming that the interaction between the drug ( A ) and the receptor ( R ) (lower figure) obeys the law of mass action, then the concentration of the drug–receptor complex ( AR ) is given by:
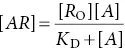
where R O= total concentration of receptors, A = agonist concentration, K D= dissociation constant and AR = concentration of occupied receptors.
As this is the equation for a hyperbola, the shape of the dose–response curve is explained if the response is directly proportional to [ AR ]. Unfortunately, this simple theory does not explain another experimental finding – some agonists, called partial agonists, cannot elicit the same maximum response as full agonists even if they have the same affinity for the receptor (top left and middle,  ). Thus, in addition to having affinity for the receptor, an agonist has another chemical property, called intrinsic efficacy, which is its ability to elicit a response when it binds to a receptor (lower figure).
). Thus, in addition to having affinity for the receptor, an agonist has another chemical property, called intrinsic efficacy, which is its ability to elicit a response when it binds to a receptor (lower figure).
A competitive antagonisthas no intrinsic efficacy and, by occupying a proportion of the receptors, effectively dilutes the receptor concentration. This causes a parallel shift of the log concentration–response curve to the right (top right,  ), but the maximum response is not depressed. In contrast, irreversible antagonistsdepress the maximum response (top right,
), but the maximum response is not depressed. In contrast, irreversible antagonistsdepress the maximum response (top right,  ). However, at low concentrations, a parallel shift of the log concentration–response curve may occur without a reduction in the maximum response (top right,
). However, at low concentrations, a parallel shift of the log concentration–response curve may occur without a reduction in the maximum response (top right,  ). Because an irreversible antagonist in effect removes receptors from the system, it is clear that not all of the receptors need to be occupied to elicit the maximum response (i.e. there is a receptor reserve).
). Because an irreversible antagonist in effect removes receptors from the system, it is clear that not all of the receptors need to be occupied to elicit the maximum response (i.e. there is a receptor reserve).
Интервал:
Закладка:
Похожие книги на «Medical Pharmacology at a Glance»
Представляем Вашему вниманию похожие книги на «Medical Pharmacology at a Glance» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Medical Pharmacology at a Glance» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.