Michael J. Neal - Medical Pharmacology at a Glance
Здесь есть возможность читать онлайн «Michael J. Neal - Medical Pharmacology at a Glance» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Medical Pharmacology at a Glance
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Medical Pharmacology at a Glance: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Medical Pharmacology at a Glance»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
is the ideal companion for all medical and healthcare students, providing a visual overview of pharmacology, and describing the basic principles of drug action, interaction, absorption, and excretion. Clear and accessible chapters organised around common diseases and conditions facilitate efficient clinical learning, and include references to drug classes and side effects, disease pathophysiology, prescribing guidelines, and more.
Now in its ninth edition, this leading guide has been thoroughly updated to reflect current guidelines and drug information. This edition features new and revised illustrations, additional pedagogical tools, and enhanced online content. Widely recognised as both the best introduction to medical pharmacology and the perfect revision tool for USMLE and pharmacology exams, this invaluable guide:
Covers a wide range of drugs used to treat conditions such as hypertension, anaemias, cancer, and affective disorders Explains drug mechanisms and the principles of drug action Discusses practical topics including drug misuse, drug indications, and side effects Includes a companion website featuring online cases, flashcards, and a list of core drugs
Medical Pharmacology at a Glance — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Medical Pharmacology at a Glance», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Drugs that are sufficiently lipid soluble to be readily absorbed orally are rapidly distributed throughout the body water compartments (  ). Many drugs are loosely bound to plasma albumin, and an equilibrium forms between the bound (PB) and free (B) drug in the plasma. Drug that is bound to plasma proteins is confined to the vascular system and cannot exert its pharmacological actions.
). Many drugs are loosely bound to plasma albumin, and an equilibrium forms between the bound (PB) and free (B) drug in the plasma. Drug that is bound to plasma proteins is confined to the vascular system and cannot exert its pharmacological actions.
If a drug is given by intravenous injection, it enters the blood and is rapidly distributed to the tissues. By taking repeated blood samples, the fall in plasma concentration of the drug with time (i.e. the rate of drug elimination) can be measured (right, top graph). Often the concentration falls rapidly at first, but then the rate of decline progressively decreases. Such a curve is called exponential, and this means that, at any given time, a constant fractionof the drug present is eliminated in unit time. Many drugs show an exponential fall in plasma concentration because the rates at which the drug elimination processes work are themselves usually proportional to the concentration of drug in the plasma. The following processes are involved.
1 Elimination in the urine by glomerular filtration (right, ).
2 Metabolism, usually by the liver.
3 Uptake by the liver and subsequent elimination in the bile ( solid line from liver).
A process that depends on the concentration at any given time is called firstorder; most drugs exhibit first‐order elimination kinetics. If any enzyme system responsible for drug metabolism becomes saturated, then the elimination kinetics change to zero order, i.e. the rate of elimination proceeds at a constant rate and is unaffected by an increased concentration of the drug (e.g. ethanol, phenytoin).
Routes of administration
Drugs can be administered orally or parenterally (i.e. by a nongastrointestinal route).
OralMost drugs are absorbed by this route and, because of its convenience, it is the most widely used. However, some drugs (e.g. benzylpenicillin, insulin) are destroyed by the acid or enzymes in the gut and must be given parenterally.
Intravenous injectionThe drug directly enters into the circulation and bypasses the absorption barriers. It is used:
where a rapid effect is required (e.g. furosemide in pulmonary oedema);
for continuous administration (infusion);
for large volumes; and
for drugs that cause local tissue damage if given by other routes (e.g. cytotoxic drugs).
Intramuscular and subcutaneous injectionsDrugs in aqueous solution are usually absorbed fairly rapidly, but absorption can be slowed by giving the drug in the form of an ester (e.g. antipsychotic depot preparations, Chapter 27).
Other routesThese include inhalation(e.g. volatile anaesthetics, some drugs used in asthma) and topical(e.g. ointments). Sublingualand rectaladministration avoids the portal circulation, and sublingual preparations in particular are valuable in administering drugs subject to a high degree of first‐pass metabolism.
Distribution and excretion
Distribution around the body occurs when the drug reaches the circulation. It must then penetrate tissues to act.
The t 1/2 (half‐life)is the time taken for the concentration of drug in the blood to fall by half its original value (right, top graph). Measurement of t 1/2allows the calculation of the elimination rate constant ( K el) from the formula:
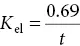
where K elis the fraction of drug present at any time that would be eliminated in unit time (e.g. K el= 0.02 min −1means that 2% of the drug present is eliminated in 1 min).
The exponential curve of plasma concentration ( C p) against time ( t ) is described by:
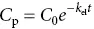
where C 0= the initial apparent plasma concentration. By taking logarithms, the exponential curve can be transformed into a more convenient straight line (right, bottom graph) from which C 0and t 1/2can readily be determined.
Volume of distribution (V D )This is the apparent volume into which the drug is distributed. Following is an intravenous injection:
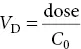
A value of V D< 5 L implies that the drug is retained within the vascular compartment. A value of <15 L suggests that the drug is restricted to the extracellular fluid, whereas large volumes of distribution ( V D> 15 L) indicate distribution throughout the total body water or concentration in certain tissues. The volume of distribution can be used to calculate the clearance of the drug.
ClearanceThis is an important concept in pharmacokinetics. It is the volume of blood or plasma cleared of drug in unit time. Plasma clearance ( Cl p) is given by the relationship:
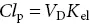
The rate of elimination = Cl p× C p. Clearance is the sum of individual clearance values. Thus, Cl p= Cl m(metabolic clearance) + Cl r(renal excretion). Clearance, but not t 1/2, provides an indication of the ability of the liver and kidney to dispose of drugs.
Drug dosageClearance values can be used to plan dosage regimens. Ideally, in drug treatment, a steady‐state plasma concentration ( C pss) is required within a known therapeutic range. A steady state will be achieved when the rate of drug entering the systemic circulation (dosage rate) equals the rate of elimination. Thus, the dosing rate = Cl × C pss. This equation could be applied to an intravenous infusion because the entire dose enters the circulation at a known rate. For oral administration, the equation becomes:
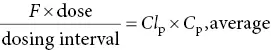
where F = bioavailability of the drug. The t 1/2value of a drug is useful in choosing a dosing interval that does not produce excessively high peaks (toxic levels) and low troughs (ineffective levels) in drug concentration.
BioavailabilityThis is a term used to describe the proportion of administered drug reaching the systemic circulation. Bioavailability is 100% following an intravenous injection ( F = 1), but drugs are usually given orally, and the proportion of the dose reaching the systemic circulation varies with different drugs and also from patient to patient. Drugs subject to a high degree of first‐pass metabolism may be almost inactive orally (e.g. glyceryl trinitrate, lidocaine).
Excretion
Renal excretionThis is ultimately responsible for the elimination of most drugs. Drugs appear in the glomerular filtrate, but if they are lipid soluble, they are readily reabsorbed in the renal tubules by passive diffusion. Metabolism of a drug often results in a less lipid‐soluble compound, aiding renal excretion (see Chapter 4).
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Medical Pharmacology at a Glance»
Представляем Вашему вниманию похожие книги на «Medical Pharmacology at a Glance» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.











Обсуждение, отзывы о книге «Medical Pharmacology at a Glance» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.