Shunichi Fukuzumi - Electron Transfer
Здесь есть возможность читать онлайн «Shunichi Fukuzumi - Electron Transfer» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Electron Transfer
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Electron Transfer: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Electron Transfer»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Electron Transfer — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Electron Transfer», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Nano‐sized charge‐separated molecules can also be obtained by using single‐walled carbon nanotubes (SWNTs) [76], which exhibit excellent chemical and physical properties as revealed by various potential applications [77–81]. Extensive efforts have so far been devoted to assemble electron donor and acceptor molecules on SWNTs [82–88]. However, the fine control of size (i.e. length) of SWNTs remains a formidable challenge, because SWNTs have seamless cylindrical structures made up of a hexagonal carbon network, which leads to the difficulty of solubilization/functionalization without treatment with strong acid or vigorous sonication [89–92]. On the other hand, the cup‐stacked carbon nanotubes (CSCNTs) that consist of cup‐shaped nanocarbon (CNC) units, which stack via van der Waals attractions, have merited special attention from the viewpoint of the conventional carbon nanotube alternatives [93–96]. The tube–tube van der Waals energy between CNCs has been counterbalanced by the thermal or photoinduced electron transfer multi‐electron reduction due to electrostatic repulsion, resulting in the highly dispersible CNCs with size homogeneity [97,98].
The CNCs with controlled size have been functionalized with a large number of porphyrin molecules [99]. The general procedure for the synthesis of the porphyrin‐functionalized cup‐shaped nanocarbons [CNC–(H 2P) n] is shown in Figure 4.7a [99]. The CNCs are first functionalized with aniline as the precursor for further functionalization with porphyrins. The aniline‐functionalized nanocarbons react with the porphyrin derivatives to construct the nanohybrids.
The structure of the CNCs of the CNC–(H 2P) nnanohybrids is shown by the TEM in Figure 4.7b, which reveals a CNC with a hollow core along the length of the nanocup with well‐controlled diameter (c. 50 nm) and size (c. 100 nm) [99]. The weight percentage of porphyrins attached to the CNCs was determined by thermogravimetric analysis (TGA) and elemental analysis to be ca. 20% [99]. This corresponds to one functional group per 640 carbon atoms of the nanocup framework for CNC–(H 2P) nnanohybrid. Thus, the π‐framework of the CNC is not destroyed despite attachment of a large number of porphyrin molecules on the CNC.
Spectroscopic evidence for the covalent functionalization of CNC–(H 2P) nnanohybrid was obtained by an intensity increase of the Raman signal at 1353 cm −1(D band) in the functionalized CNC as compared with the pristine CSCNTs [99], because the D band has been used for monitoring the process of functionalization that transforms sp 2to sp 3sites [99]. The UV–vis absorption spectrum of CNC–(H 2P) nnanohybrid agreed with that of the superposition of ref‐H 2P [tetrakis( N ‐octadecyl‐4‐aminocarboxyphenyl)porphyrin] and CNCs, indicating that there is no significant interaction between attached porphyrins and CSCNTs in the ground states [99].
The fluorescence lifetime of CNC–(H 2P) nwas determined to be 3.0 ± 0.1 ns, which is much shorter than that of ref‐H 2P (14.1 ± 0.1 ns) [99]. The fluorescence emission at 650 nm was also quenched in CNC–(H 2P) n[99]. The short fluorescence lifetime of CNC–(H 2P) nand an efficient fluorescence quenching of porphyrins in CNC–(H 2P) nas compared to the ref‐H 2P may result from the photoinduced electron transfer from the singlet excited state of H 2P ( 1H 2P *) to CNC in CNC–(H 2P) n. The occurrence of photoinduced electron transfer to afford the charge‐separated (CS) state of CNC–(H 2P) nwas confirmed by nanosecond laser flash photolysis measurements in Figure 4.8, where the absorption bands in the visible and near infrared (NIR) regions are attributed to H 2P ·+, which are clearly different from the triplet–triplet absorption of ref‐H 2P [99]. The formation of the CS state was also confirmed by EPR measurements under photoirradiation of CNC–(H 2P) nin frozen N , N ‐diemthylformamide (DMF) at 153 K. The observed isotropic EPR signal at g = 2.0044 agrees with that of ref‐H 2P ·+produced by one‐electron oxidation with [Ru(bpy) 3] 3+(bpy = 2,2′‐bipyridine) in deaerated CHCl 3[99]. The EPR signal corresponding to the reduced carbon‐based nanomaterials was too broad to be detected, probably due to delocalization of electrons in CNC [99].
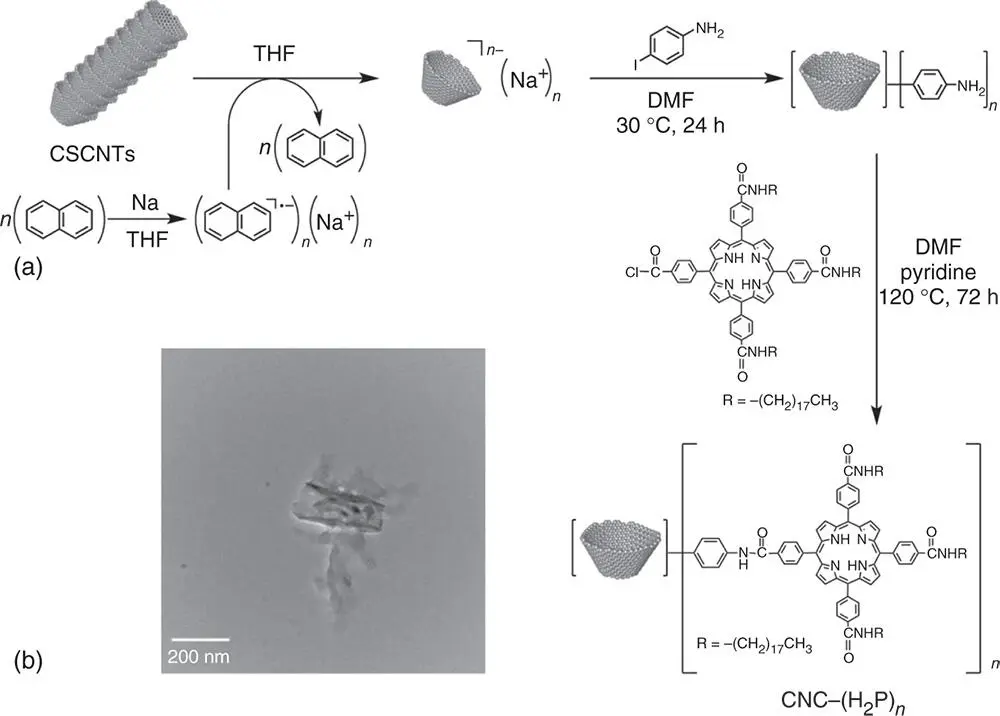
Figure 4.7(a) Synthetic procedure of CNC–(H 2P) n. (b) TEM image of CNC–(H 2P) n.
Source: Ohtani et al. 2009 [99]. Reproduced with permission of John Wiley & Sons.
The CS state of CNC–(H 2P) ndetected in Figure 4.8a decays obeying clean first‐order kinetics: the first‐order plots at different initial CS concentrations afford linear correlations with the same slope (Figure 4.8b) [99]. Thus, the decay of the CS state results from back electron transfer in the nanohybrid rather than intermolecular back electron transfer from CNC ·−to H 2P ·+. The CS lifetime was determined from the first‐order plots in Figure 4.8b to be 0.64 ± 0.01 ms, which is the longest lifetime ever reported for electron donor‐attached nanocarbon materials [99]. Such a long CS lifetime may be ascribed to the efficient electron migration in the CNCs following CS.
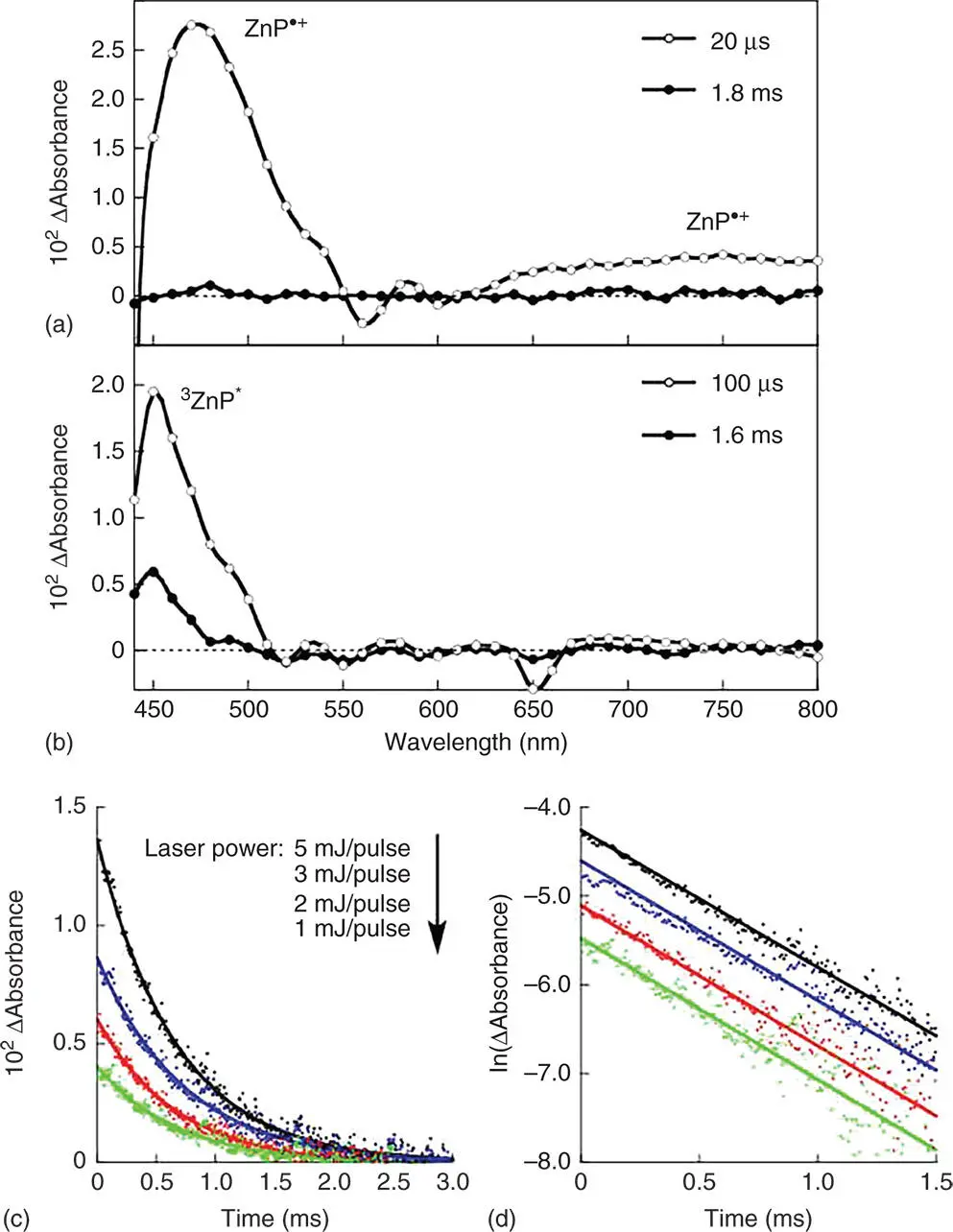
Figure 4.8(a) Transient absorption spectra of (a) CNC–(H 2P) ntaken at 20 and 1.8 ms after laser excitation at 426 nm and (b) ref‐H 2P in deaerated DMF at 298 K taken at 100 ms and 1.6 ms after laser excitation at 426 nm. (c) Decay time profiles and (d) first‐order plots at 470 nm with different laser powers (5, 3, 2, and 1 mJ/pulse).
Source: Ohtani et al. 2009 [99]. Reproduced with permission of John Wiley & Sons.
Конец ознакомительного фрагмента.
Текст предоставлен ООО «ЛитРес».
Прочитайте эту книгу целиком, на ЛитРес.
Безопасно оплатить книгу можно банковской картой Visa, MasterCard, Maestro, со счета мобильного телефона, с платежного терминала, в салоне МТС или Связной, через PayPal, WebMoney, Яндекс.Деньги, QIWI Кошелек, бонусными картами или другим удобным Вам способом.
Интервал:
Закладка:
Похожие книги на «Electron Transfer»
Представляем Вашему вниманию похожие книги на «Electron Transfer» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Electron Transfer» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.