Shunichi Fukuzumi - Electron Transfer
Здесь есть возможность читать онлайн «Shunichi Fukuzumi - Electron Transfer» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Electron Transfer
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Electron Transfer: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Electron Transfer»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Electron Transfer — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Electron Transfer», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
Covalently and non‐covalently linked porphyrin–quinone dyads constitute one of the most extensively investigated photosynthetic reaction center models, in which the fast photoinduced electron transfer from the porphyrin singlet excited state to the quinone occurs to produce the CS state, mimicking well the photosynthetic electron transfer [52–54]. Unfortunately, the CR rates of the CS state of porphyrin–quinone dyads are also fast and the CS lifetimes are mostly on the order of picoseconds or subnanoseconds in solution [52–54]. In general, a three‐dimensional C 60is superior to a two‐dimensional quinone in terms of the smaller reorganization of electron transfer of C 60as compared with quinone (vide supra) to attain the long‐lived CS state [31–33,55]. When the geometry between a porphyrin ring and quinone is optimized by using hydrogen bonds, which can also control the redox potentials of quinones, however, a surprisingly long lifetime up to one microsecond has been attained [56]. In a series of ZnP– n –Q ( n = 3, 6, 10) in Scheme 4.3, the hydrogen bond between two amide groups provides a structural scaffold to assemble the donor (ZnP) and the acceptor (Q) moiety, leading to attaining the long‐lived CS state [56].
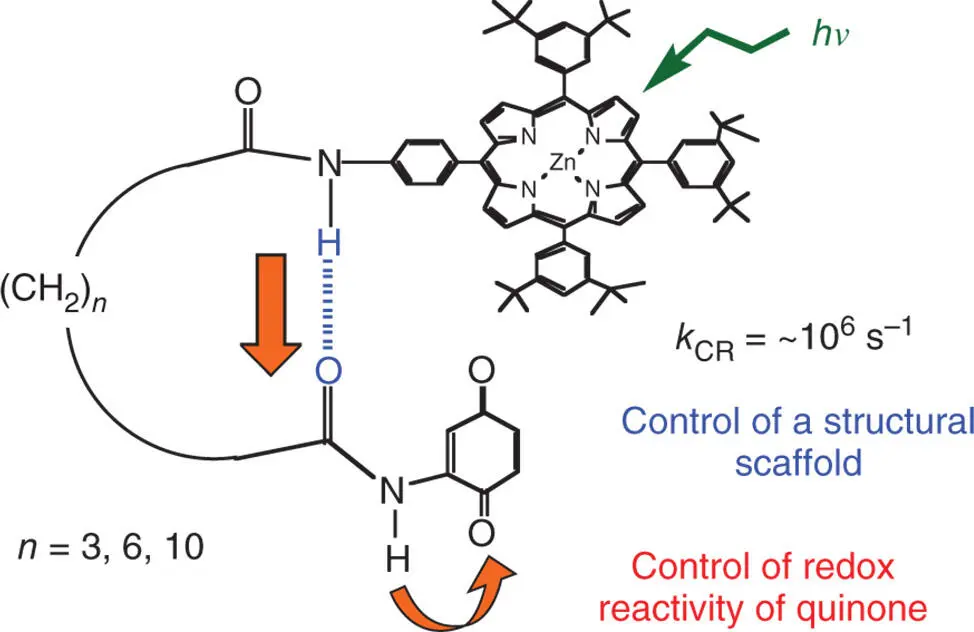
Scheme 4.3Zinc porphyrin–quinone linked dyads (ZnP– n –Q; n = 3, 6, 10) with hydrogen bonds.
Source: Okamoto and Fukuzumi 2005 [56]. Reproduced with permission of American Chemical Society.
As described above, the closely linked donor–acceptor dyads afford long‐lived CS states. As long as porphyrins and C 60are used as components of donor–acceptor dyads, however, the low lying triplet energies of porphyrins and C 60have precluded to attain the long‐lived CS states with a higher energy than the triplet energies [35]. In such a case, it is highly desired to find a chromophore that has a high triplet energy and a small λ value of electron transfer. Among many choromophores, acridinium ion is the best candidate for such a purpose, since the λ value for the electron self‐exchange between the acridinium ion and the corresponding one‐electron reduced radical (acridinyl radical) is the smallest (0.3 eV) among the redox‐active organic compounds [57]. Another important property of acridinium ion is a high triplet excited energy [58,59]. Thus, an electron donor moiety (mesityl group) is directly connected at the 9‐position of the acridinium ion to yield 9‐mesityl‐10‐methylacridinium ion (Acr +–Mes) [60], in which the solvent reorganization of electron transfer is minimized because of the short linkage between the donor and acceptor moieties. The X‐ray crystal structure of Acr +–Mes is shown in Figure 4.2a [60]. The dihedral angle made by aromatic ring planes is perpendicular and therefore there is no π conjugation between the donor and acceptor moieties. Indeed, the absorption and fluorescence spectra of Acr +–Mes are superpositions of the spectra of each component, i.e. mesitylene and 10‐methylacridinium ion. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) orbitals of Acr +–Mes calculated by a density functional theory (DFT) method with Gaussian 98 (B3LYP/6‐31G* basis set) are localized on mesitylene and acridinium moieties (Figure 4.2b,c), respectively [60]. The energy of the electron‐transfer state (Acr ·–Mes ·+) in PhCN is determined by the redox potentials of each component of Acr +–Mes as 2.37 eV [60].
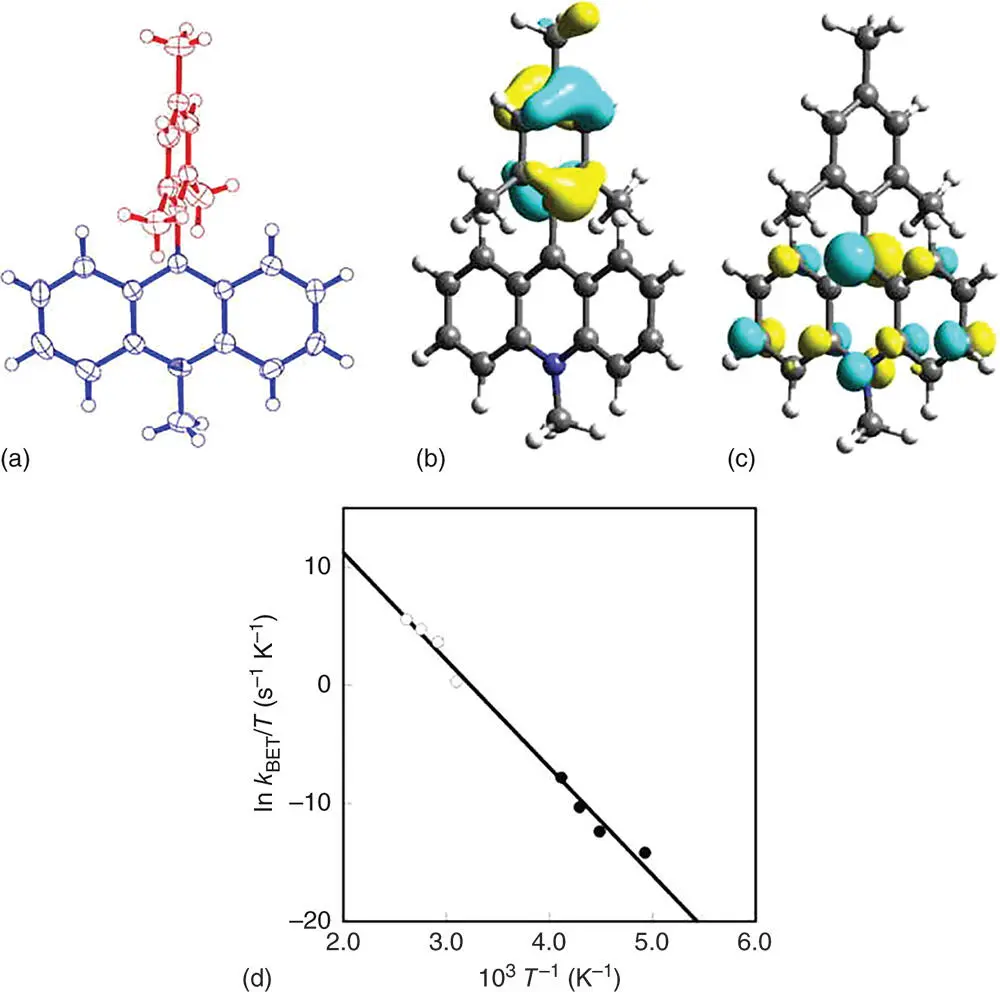
Figure 4.2(a) X‐ray crystal structure of Acr +–Mes. (b) HOMO and (c) LUMO orbitals calculated by DFT method with Gaussian 98 (B3LYP/6‐31G* basis set). (d) Plot of k BET/ T vs. T −1.
Source: Fukuzumi et al. 2004 [60]. Reproduced with permission of American Chemical Society.
Photoirradiation of a deaerated PhCN solution of Acr +–Mes by a nanosecond laser light at 430 nm results in the formation of Acr ·–Mes ·+with a quantum yield close to unity (98%) via photoinduced electron transfer from the mesitylene moiety to the singlet excited state of the acridinium ion moiety ( 1Acr +*–Mes) [60]. The intramolecular back electron transfer from the Acr ·moiety to the Mes ·+moiety in Acr ·–Mes ·+was too slow to compete with the intermolecular transfer ( k BET) in Figure 4.2d, agreeing with the Marcus equation in the deeply inverted region (Eq. (2.1)). The lifetime of the electron‐transfer state in frozen medium becomes longer with decreasing temperature to approach a virtually infinite value at 77 K [60]. However, the decay time profile of Acr ·–Mes ·+in solution obeyed second‐order kinetics (NOT first‐order kinetics) [60]. This is the same as the case of Fc +–ZnP–H 2P–C 60 ·−, in which bimolecular back electron transfer predominates due to the slow intramolecular back electron transfer (vide supra) [39]. In contrast, the decay of Acr ·–Mes ·+obeys first‐order kinetics in PhCN at high temperatures [60]. This indicates that the rate of the intramolecular back electron transfer of Acr ·–Mes ·+becomes much faster than the rate of the intermolecular back electron transfer at higher temperatures because of the larger activation energy of the former than that of the latter. Such a remarkable result has sparked a flurry of work by others in the field of artificial photosynthesis [61].
Benniston et al. claimed that the triplet excited state of the acridinium ion moiety ( 3Acr +*–Mes) might be formed rather than the electron‐transfer state (Acr ·–Mes ·+) and that the energy of 3Acr +*–Mes is lower than that of Acr ·–Mes ·+[62]. They reported that the triplet excitation energy of Acr +–Mes was 1.96 eV based on the phosphorescence spectrum [62]. If this value were correct, the one‐electron oxidation potential ( E ox) of 3Acr +*–Mes would be −0.08 V vs. saturated calomel electrode (SCE), which is determined from the one‐electron oxidation potential of the Mes moiety (1.88 V) [60] and the triplet excitation energy (1.96 V). In such a case, electron transfer from the triplet excited state of Acr +–Mes to N , N ‐dihexylnaphthalenediimide (NIm: E red= –0.46 V vs. SCE) would be energetically impossible judging from the positive free energy change of electron transfer (0.38 eV). However, the addition of NIm (1.0 × 10 −3M) to a PhCN solution of Acr +–Mes and laser photoexcitation results in the formation of NIm ·−as detected clearly by the well‐known absorption bands at 480 and 720 nm [63,64], accompanied by the decay of transient absorption at 510 nm due to the Acr ·moiety of the electron‐transfer (ET) state as shown in Figure 4.3a [65]. Similarly, the addition of aniline (3.0 × 10 −5M) to a PhCN solution of Acr +–Mes results in the formation of aniline radical cation ( λ max= 430 nm) [66], accompanied by decay of the Mes ·+moiety at 500 nm as shown in Figure 4.3b [65]. The rate constant of formation of aniline radical cation was determined to be 5.6 × 10 9M −1s −1, which is close to the diffusion rate constant in PhCN [60]. Thus, the photogenerated state of Acr +–Mes has both the reducing and oxidizing abilities to reduce NIm and to oxidize aniline, respectively. Only the electron‐transfer state (Acr ·–Mes ·+) has such a dual ability, which has now been well confirmed for electron‐transfer oxidation of many electron donors with Acr ·–Mes ·+and electron‐transfer reduction of many electron acceptors such as hexyl viologen, p ‐benzoquinone, and Selectfluor (fluorinating reagent) with Acr ·–Mes ·+[67–70]. However, this conclusion is contradictory to the reported triplet energy (1.96 eV), which is lower in energy than the ET state [62]. This contradiction comes from an acridine impurity, which may be left in the preparation of by Benniston et al. who synthesized the compound via methylation of the corresponding acridine [62]. The yield of acridinium ion is about 50–70% after reflux at high temperature for a few days [62]. In such a case, acridine may remain as an impurity even after purification of the acridinium ion by recrystallization. When Acr +–Mes was prepared by the Grignard reaction of 10‐methyl‐9(10 H )acridone with 2‐mesitylmagnesium bromide, there was no acridine [60]. Thus, Acr +–Mes without acridine afforded no phosphorescence spectrum in both deaerated glassy 2‐MeTHF and ethanol at 77 K. It is well known that acridine derivatives exhibit phosphorescence at 15650–15850 cm −1[71]. It was confirmed that the phosphorescence maximum of 9‐phenylacridine in glassy 2‐MeTHF at 77 K afforded the same spectrum reported by Benniston et al. [62] Thus, the reported low triplet energy of Acr +–Mes, which contradicts our results on the long‐lived electron‐transfer state, results from the acridine impurity contained in Acr +–Mes used by Benniston et al. who also reported that photoirradiation of a PhCN solution of Acr +–Mes results in the formation of the acridinyl radical (Acr ·–Mes) [62]. They implied that this stable radical species could be mistaken as a long‐lived electron‐transfer state [62]. When PhCN is purified, however, no change in the absorption spectrum is observed [60,65]. The formation of Acr ·–Mes results from electron transfer from a donor impurity contained in unpurified PhCN (e.g. aniline) to the Mes ·+moiety of Acr ·–Mes ·+as indicated in Figure 4.3b. Even an extremely small amount (5.0 × 10 −5M) of aniline is enough to react with Acr ·–Mes ·+to produce Acr ·–Mes, which is stable due to the bulky Mes substituent, because the lifetime of Acr ·–Mes ·+is long enough to react with such a small concentration of an electron donor. It should be noted that no net photochemical reaction occurs without a donor impurity because the long‐lived Acr ·–Mes ·+decays via bimolecular back electron transfer to the ground state [60,65]. Thus, misleading effects of impurities indeed result from the long‐lived electron‐transfer state, which has both oxidizing and reducing abilities.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Electron Transfer»
Представляем Вашему вниманию похожие книги на «Electron Transfer» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Electron Transfer» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.