Shunichi Fukuzumi - Electron Transfer
Здесь есть возможность читать онлайн «Shunichi Fukuzumi - Electron Transfer» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Electron Transfer
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Electron Transfer: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Electron Transfer»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Electron Transfer — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Electron Transfer», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:

Figure 4.3Transient absorption spectra of Acr +–Mes (5.0 × 10 −5M) in deaerated MeCN at 298 K taken at 2 and 20 μs after laser excitation at 430 nm in the presence of (a) N,N ‐dihexylnaphthalenediimide (1.0 × 10 −3M) or (b) aniline (3.0 × 10 −5M). Inset: Time profiles of the absorbance decay at 510 nm and the rise at 720 nm and (b) the decay at 500 nm and the rise at 430 nm.
Source: Fukuzumi and coworkers 2005 [65]. Reproduced with permission of Royal Society of Chemistry.
In contrast to the photoirradiation of a purified PhCN solution of Acr +–Mes at 298 K, which results in no change in the absorption spectrum (Figure 4.4a), when the photoirradiation of the same solution was performed at low temperatures (213–243 K) with a 1000 W high‐pressure mercury lamp through the UV light cutting filter (>390 nm) and the sample was cooled to 77 K, the color of the frozen sample at 77 K was clearly changed as shown in the inset of Figure 4.4b. When a glassy 2‐methyltetrahydrofuran (2‐MeTHF) is employed for the photoirradiation of Acr +–Mes at low temperature, the resulting glassy solution measured at 77 K affords the absorption spectrum due to the electron‐transfer state, which consists of the absorption bands of the Acr ·moiety (500 nm) and the Mes ·+moiety (470 nm) as shown in Figure 4.4b. No decay of the absorption due to the electron‐transfer state in Figure 4.2b was observed until liquid nitrogen ran out [65].
The long lifetime of the ET state of Acr +–Mes has allowed observing the structural change in the Acr +–Mes(ClO 4 −) crystal upon photoinduced ET directly by using laser pump and X‐ray probe crystallographic analysis (Figure 4.5) [72]. Upon photoexcitation of the crystal of Acr +–Mes(ClO 4 −), the N ‐methyl group of the Acr +moiety was bent and its bending angle was 10.3(16)° when the N ‐methyl carbon moved 0.27(4) Å away from the mean plane of the ring as shown in Figure 4.5[72]. This bending is caused by the photoinduced electron transfer from the Mes moiety to the Acr +moiety to produce Acr ·–Mes ·+, because the sp 2carbon of the N ‐methyl group of Acr +is changed to the sp 3carbon in the one‐electron reduced state (Acr ·) [72]. The bending of the N ‐methyl group by photoexcitation was accompanied by the rotation and movement of the ClO 4 −by the electrostatic interaction with the Mes ·+moiety (Figure 4.5) [72]. Thus, the observed bending of the N ‐methyl group and the movement of ClO 4 −provide strong evidence for the generation of the ET state of Acr +–Mes upon photoexcitation. In contrast to the case of Acr +–Mes, no geometrical difference was observed upon photoexcitation of Acr +–Ph, which does not afford the ET state [72].
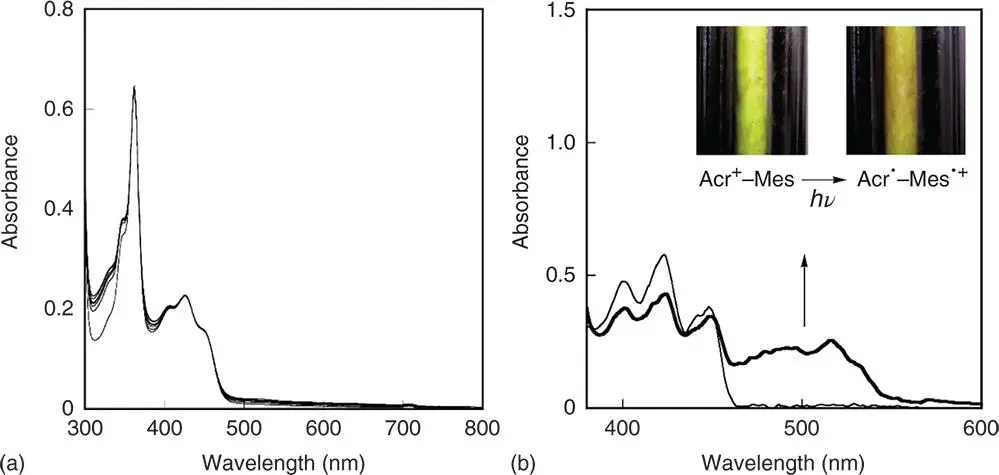
Figure 4.4(a) UV–vis spectral change in the steady‐state photolysis of a deaerated PhCN solution of Acr +–Mes (3.3 × 10 –5M). Spectra were recorded at 90‐second interval. (b) UV–vis absorption spectra obtained by photoirradiation with high‐pressure mercury lamp of deaerated 2‐MeTHF glasses of Acr +–Mes at 77 K. Inset: picture images of frozen PhCN solutions of Acr +–Mes before and after photoirradiation at low temperatures and taken at 77 K.
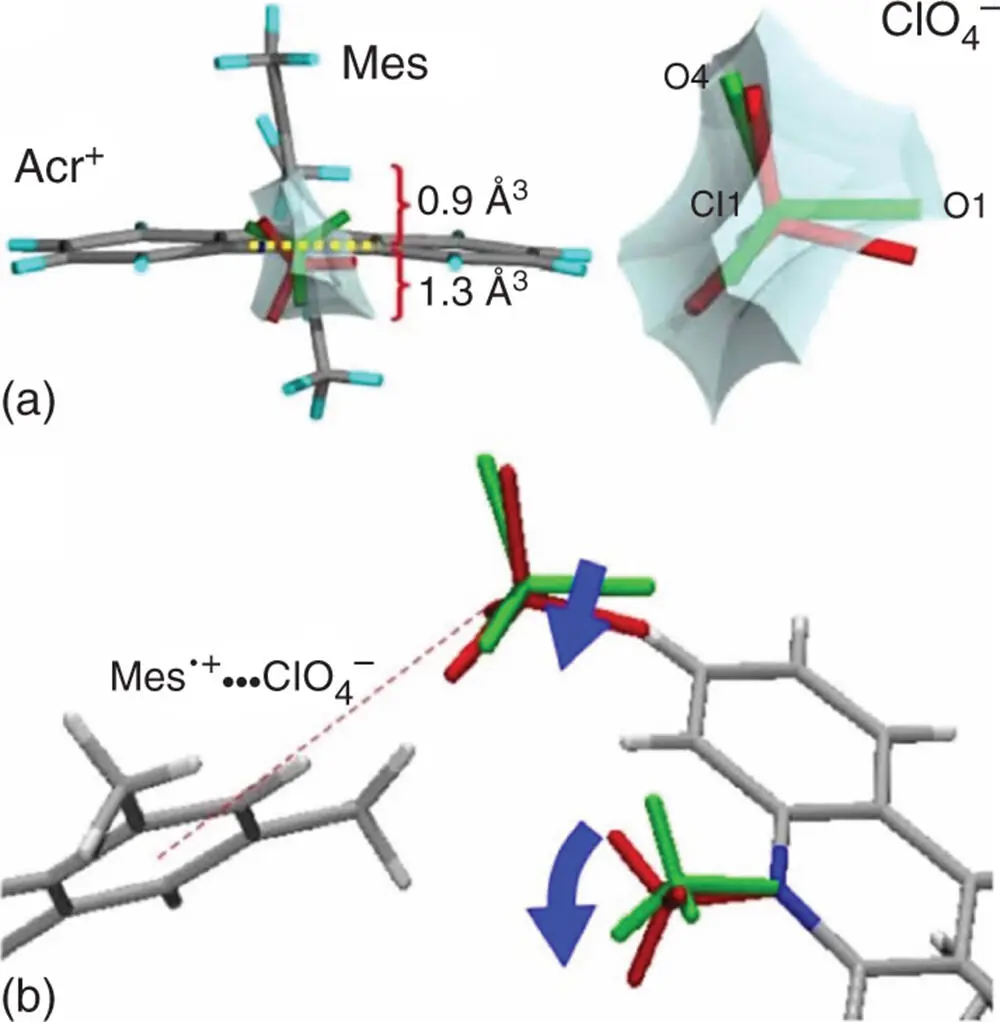
Figure 4.5(a) Diagram of the reaction cavity: (left) diagram around the N ‐methyl group, with numbers indicating the volumes of the divided cavity formed by the dotted line; (right) drawing around ClO 4 −. (b) Cooperative photoinduced geometrical changes. The dashed line indicates the suggested Mes ·+⋯ClO 4 −electrostatic interaction.
Source: Hoshino et al. 2012 [72]. Reproduced with permission of American Chemical Society.
Immobilization of Acr +–Mes has also been achieved by incorporating Acr +–Mes cation into nanosized mesoporous silica–alumina (AlMCM‐41), which has cation exchange sites to obtain a nanocomposite (Acr +–Mes@AlMCM‐41) [73]. The shape and size of nanosized AlMCM‐41 were controlled by changing the preparation conditions as shown in Figure 4.6, where TEM images reveal a tubular or rod‐like (tAlMCM‐41) morphology in the diameter of 50–100 nm with the length of 0.2–2 μm array (part a) and also a sphere morphology (sAlMCM‐41, part b) [73]. The X‐ray powder pattern of tAlMCM‐41 exhibited a well‐resolved pattern with a prominent peak (100) observed at c. 2 θ = 2.56°, indicating a highly ordered material with a hexagonal array [73]. Uniform channels c. 4 nm in diameter exist in a tube. Because the Acr +–Mes molecular size is small enough as compared with the pore size of mesoporous silica with its diameter of more than 3 nm, cation exchange with Acr +–Mes occurs spontaneously upon mixing Na +–exchanged AlMCM‐41 with Acr +–Mes in acetonitrile [73]. The cation exchange percentages of tAlMCM‐41 and sAlMCM‐41 by Acr +–Mes were determined to be 16% and 18%, respectively [73]. The Acr +–Mes incorporated into AlMCM‐41 is stable without leaching out in acetonitrile at room temperature [73].
Upon photoexcitation of Acr +–Mes@tAlMCM‐41 suspended in MeCN, photoinduced ET from the Mes moiety to the singlet excited state of the Acr +moiety occurred within 10 ps to produce the ET state as detected by laser flash photolysis and electron paramagnetic resonance (EPR) measurements [60,67]. In contrast to the case in solution (vide supra), no bimolecular decay of the ET state occurs because each Acr +–Mes molecule is isolated inside AlMCM‐41 [73]. The lifetime of the ET state of Acr +–Mes@tAlMCM‐41 suspended in acetonitrile was determined to be 2.3 seconds at 198 K, which is much longer than that in solution because of the inhibition of bimolecular BET in AlMCM‐41 as illustrated in Figure 4.6[73]. Thus, incorporation of a simple electron donor–acceptor dyad into AlMCM‐41 has made it possible to elongate the lifetime of the charge‐separated state, which is longer than that of the bacterial photosynthetic reaction center (one second) [74].
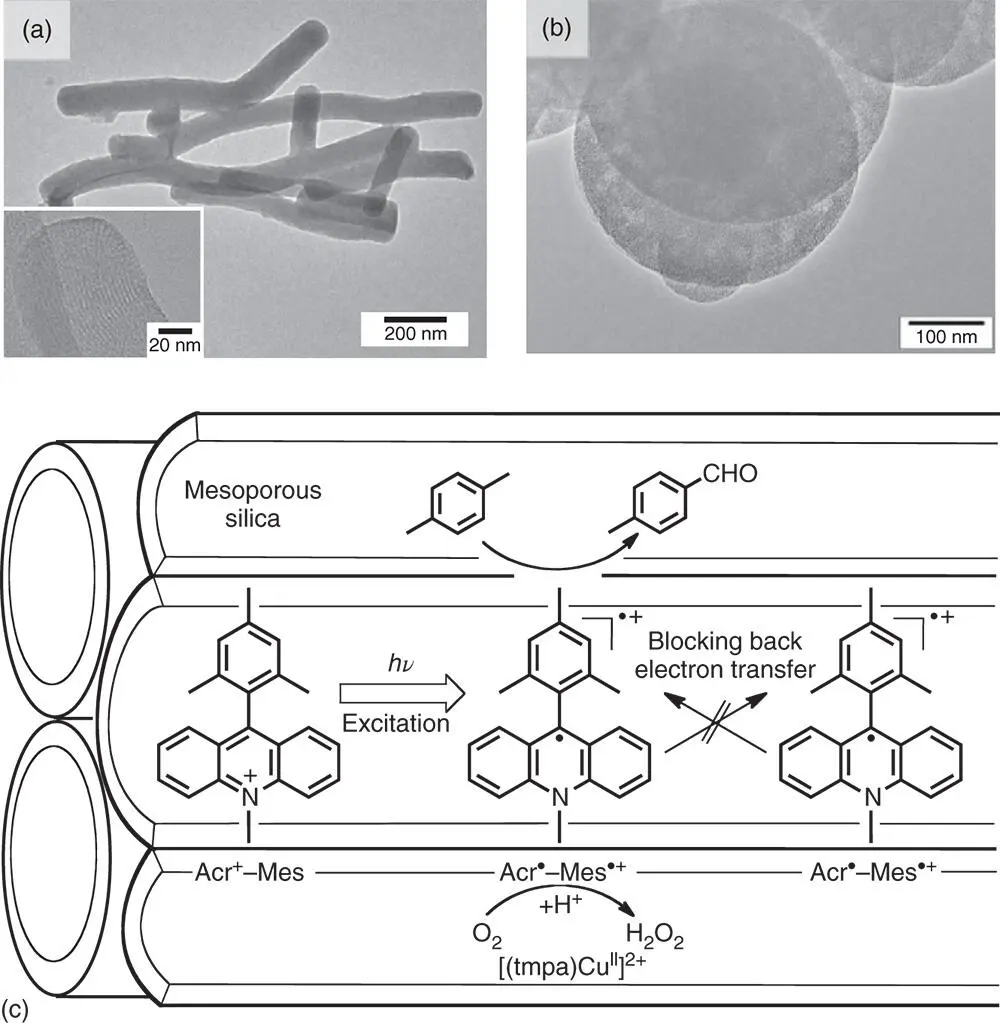
Figure 4.6Transmission electron microscope (TEM) images of (a) tAlMCM‐41 and (b) sAlMCM‐41 (the high‐resolution image of tAlMCM‐41 is inserted in (a)). (c) Reaction scheme of photocatalytic oxygenation of p ‐xylene with Acr +–Mes and [(tmpa)Cu II] 2+incorporated into sAlMCM‐41.
Source: Fukuzumi et al. 2012 [73]. Reproduced with permission of PNAS.
The triplet ET state of Acr ·–Mes ·+@tAlMCM‐41 was detected by an EPR spectrum measured at 4 K, which exhibited a fine structure together with a strong sharp signal at g = 4.0 [73]. The distance between two electron spins was determined from the zero‐field splitting parameters to be 7.7 Å, which agrees with the expected distance of 7.2 Å between an sp 2carbon atom at the 4 position of the mesityl moiety and sp 2carbon atoms at the 3 and 6 positions of the acridinyl moiety [73]. Polycondensation of Acr +–Mes‐bridged organosilane in the presence of a nonionic surfactant is also reported to yield a mesostructured organosilica solid with a functional framework that exhibited long‐lived photoinduced CS [75].
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Electron Transfer»
Представляем Вашему вниманию похожие книги на «Electron Transfer» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.












Обсуждение, отзывы о книге «Electron Transfer» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.