Bioinformatics
Здесь есть возможность читать онлайн «Bioinformatics» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:Bioinformatics
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:4 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
Bioinformatics: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «Bioinformatics»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
“This book is a gem to read and use in practice.”
— "This volume has a distinctive, special value as it offers an unrivalled level of details and unique expert insights from the leading computational biologists, including the very creators of popular bioinformatics tools."
— “A valuable survey of this fascinating field. . . I found it to be the most useful book on bioinformatics that I have seen and recommend it very highly.”
— “This should be on the bookshelf of every molecular biologist.”
— The field of bioinformatics is advancing at a remarkable rate. With the development of new analytical techniques that make use of the latest advances in machine learning and data science, today’s biologists are gaining fantastic new insights into the natural world’s most complex systems. These rapidly progressing innovations can, however, be difficult to keep pace with.
The expanded fourth edition of the best-selling
aims to remedy this by providing students and professionals alike with a comprehensive survey of the current field. Revised to reflect recent advances in computational biology, it offers practical instruction on the gathering, analysis, and interpretation of data, as well as explanations of the most powerful algorithms presently used for biological discovery.
offers the most readable, up-to-date, and thorough introduction to the field for biologists at all levels, covering both key concepts that have stood the test of time and the new and important developments driving this fast-moving discipline forwards.
This new edition features:
New chapters on metabolomics, population genetics, metagenomics and microbial community analysis, and translational bioinformatics A thorough treatment of statistical methods as applied to biological data Special topic boxes and appendices highlighting experimental strategies and advanced concepts Annotated reference lists, comprehensive lists of relevant web resources, and an extensive glossary of commonly used terms in bioinformatics, genomics, and proteomics
is an indispensable companion for researchers, instructors, and students of all levels in molecular biology and computational biology, as well as investigators involved in genomics, clinical research, proteomics, and related fields.
Bioinformatics — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «Bioinformatics», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
2 Frequency. In the same way that amino acid residues cannot freely substitute for one another, the matrices also need to reflect how often particular residues occur among the entire constellation of proteins. Residues that are rare are given more weight than residues that are more common.
3 Evolution. By design, scoring matrices implicitly represent evolutionary patterns, and matrices can be adjusted to favor the detection of closely related or more distantly related proteins. The choice of matrices for different evolutionary distances is discussed below.
There are also subtle nuances that go into constructing a scoring matrix, and these are described in an excellent review by Henikoff and Henikoff (2000).
How these various factors are actually represented within a scoring matrix can be best demonstrated by deconstructing the most commonly used scoring matrix, called BLOSUM62 ( Figure 3.1). Each of the 20 amino acids (as well as the standard ambiguity codes) is shown along the top and down the side of a matrix. The scores in the matrix actually represent the logarithm of an odds ratio ( Box 3.1) that considers how often a particular residue is observed, in nature, to replace another residue. The odds ratio also considers how often a particular residue would be replaced by another if replacements occurred in a random fashion (purely by chance). Given this, a positive score indicates two residues that are seen to replace each other more often than by chance, and a negative score indicates two residues that are seen to replace each other less frequently than would be expected by chance. Put more simply, frequently observed substitutions have positive scores and infrequently observed substitutions have negative scores.
Box 3.1Scoring Matrices and the Log Odds Ratio
Protein scoring matrices are derived from the observed replacement frequencies of amino acids for one another. Based on these probabilities, the scoring matrices are generated by applying the following equation:
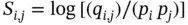
where p iis the probability with which residue i occurs among all proteins and p jis the probability with which residue j occurs among all proteins. The quantity q i,jrepresents how often the two amino acids i and j are seen to align with one another in multiple sequence alignments of protein families or in sequences that are known to have a biological relationship. Therefore, the log odds ratio S i,j(or “lod score”) represents the ratio of observed vs. random frequency for the substitution of residue i by residue j . For commonly observed substitutions, S i,jwill be greater than zero. For substitutions that occur less frequently than would be expected by chance, S i,jwill be less than zero. If the observed frequency and the random frequency are the same, S i,jwill be zero.
To explain the meaning of the numbers in the matrix more fully, imagine that two sequences have been aligned with one another, and it is now necessary to assess how well a residue in sequence A matches to a residue in sequence B at any given position of the alignment. Using the scoring matrix in Figure 3.1as our starting point,
The values on the diagonal represent the score that would be conferred for an exact match at a given position, and these numbers are always positive. So, if a tryptophan residue (W) in sequence A is aligned with a tryptophan residue in sequence B, this match would be conferred 11 points, the value where the row marked W intersects the column marked W. Also notice that 11 is the highest value on the diagonal, so the high number of points assigned to a W:W alignment reflects not only the exact match but also the fact that tryptophan is the rarest of amino acids found in proteins. Put otherwise, the W:W alignment is much less likely to occur in general and, in turn, is more likely to be correct.
Moving off the diagonal, consider the case of a conservative substitution: a tyrosine (Y) for a tryptophan. The intersection of the row marked Y with the column marked W yields a value of 2. The positive value implies that the substitution is observed to occur more often in an alignment than it would by chance, but the replacement is not as good as if the tryptophan residue had been preserved (2 < 11) or if the tyrosine residue had been preserved (2 < 7).
Finally, consider the case of a non-conservative substitution: a valine (V) for a tryptophan. The intersection of the row marked V with the column marked W yields a value of −3. The negative value implies that the substitution is not observed to occur frequently and may arise more often than not by chance.
Although the meaning of the numbers and relationships within the scoring matrices seems straightforward enough, some value judgments need to be made as to what actually constitutes a conservative or non-conservative substitution and how to assess the frequency of either of those events in nature. This is the major factor that differentiates scoring matrices from one another. To help the reader make an intelligent choice, a discussion of the approach, advantages, and disadvantages of the various available matrices is in order.
PAM Matrices
The first useful matrices for protein sequence analysis were developed by Dayhoff et al. (1978). The basis for these matrices was the examination of substitution patterns in a group of proteins that shared more than 85% sequence identity. The analysis yielded 1572 changes in the 71 groups of closely related proteins that were examined. Using these results, tables were constructed that indicated the frequency of a given amino acid substituting for another amino acid at a given position.
As the sequences examined shared such a high degree of similarity, the resulting frequencies represent what would be expected over short evolutionary distances. Further, given the close evolutionary relationship between these proteins, one would expect that the observed mutations would not significantly change the function of the protein. This is termed acceptance : changes that can be accommodated through natural selection and result in a protein with the same or similar function as the original. As individual point mutations were considered, the unit of measure resulting from this analysis is the point accepted mutation or PAM unit. One PAM unit corresponds to one amino acid change per 100 residues, or roughly 1% divergence.
Several assumptions went into the construction of the PAM matrices. One of the most important assumptions was that the replacement of an amino acid is independent of previous mutations at the same position. Based on this assumption, the original matrix was extrapolated to come up with predicted substitution frequencies at longer evolutionary distances. For example, the PAM1 matrix could be multiplied by itself 100 times to yield the PAM100 matrix, which would represent what one would expect if there were 100 amino acid changes per 100 residues. (This does not imply that each of the 100 residues has changed, only that there were 100 total changes; some positions could conceivably change and then change back to the original residue.) As the matrices representing longer evolutionary distances are an extrapolation of the original matrix derived from the 1572 observed changes described above, it is important to remember that these matrices are, indeed, predictions and are not based on direct observation. Any errors in the original matrix would be exaggerated in the extrapolated matrices, as the mere act of multiplication would magnify these errors significantly.
Читать дальшеИнтервал:
Закладка:
Похожие книги на «Bioinformatics»
Представляем Вашему вниманию похожие книги на «Bioinformatics» списком для выбора. Мы отобрали схожую по названию и смыслу литературу в надежде предоставить читателям больше вариантов отыскать новые, интересные, ещё непрочитанные произведения.




Обсуждение, отзывы о книге «Bioinformatics» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.