2D Monoelements
Здесь есть возможность читать онлайн «2D Monoelements» — ознакомительный отрывок электронной книги совершенно бесплатно, а после прочтения отрывка купить полную версию. В некоторых случаях можно слушать аудио, скачать через торрент в формате fb2 и присутствует краткое содержание. Жанр: unrecognised, на английском языке. Описание произведения, (предисловие) а так же отзывы посетителей доступны на портале библиотеки ЛибКат.

- Название:2D Monoelements
- Автор:
- Жанр:
- Год:неизвестен
- ISBN:нет данных
- Рейтинг книги:5 / 5. Голосов: 1
-
Избранное:Добавить в избранное
- Отзывы:
-
Ваша оценка:
2D Monoelements: краткое содержание, описание и аннотация
Предлагаем к чтению аннотацию, описание, краткое содержание или предисловие (зависит от того, что написал сам автор книги «2D Monoelements»). Если вы не нашли необходимую информацию о книге — напишите в комментариях, мы постараемся отыскать её.
Recent advances in phosphorene The diverse properties of two-dimensional antimonene, of graphene and its derivatives The molecular docking simulation study used to analyze the binding mechanisms of graphene oxide as a cancer drug carrier Metal-organic frameworks (MOFs)-derived carbon (graphene and carbon nanotubes) and MOF-carbon composite materials, with a special emphasis on the use of these nanostructures for energy storage devices (supercapacitors) Two-dimensional monoelements classification like graphene application in field-effect transistors for sensing and biosensing Graphene-based ternary materials as a supercapacitor electrode Rise of silicene and its applications in gas sensing
2D Monoelements — читать онлайн ознакомительный отрывок
Ниже представлен текст книги, разбитый по страницам. Система сохранения места последней прочитанной страницы, позволяет с удобством читать онлайн бесплатно книгу «2D Monoelements», без необходимости каждый раз заново искать на чём Вы остановились. Поставьте закладку, и сможете в любой момент перейти на страницу, на которой закончили чтение.
Интервал:
Закладка:
In contrast to pure phosphorene, the first peak of the optical absorption in all P 4O 2structures is characterized by a dark exciton with long-lifetime. This result makes these new systems promising candidates as molecular sensors or applications in on-chip communication. Studying the excitonic effects of half-oxidized phosphorene conformers reveals that the wave function in the dangling phosphorene extends along the armchair direction, which is similar to pure phosphorene (see Figure 1.12). These results indicate that six half-oxidized phosphorene conformers are potential candidates for electronic devices and photovoltaic applications [20].
1.3.2.3 Strain Effect
Besides the high flexibility and strong anisotropic elastic properties of phosphorene, oxidation is so important to tune its elastic properties and extending the corresponding applications. Phosphorene half-oxides can be stretched becoming ideal for devices requiring flexibility [79]. Moreover, the degree of oxidation influences significantly the elastic parameters [30–80]. The polar plot of Young modulus and Poisson ratios reveals that the maximal values are attempted for armchair-strain resulting super flexible structures. However, it is hard to implement zigzag deformation direction that shows minimal values of elastic parameters (see Figures 1.13a, b). Importantly, the Poisson ratios of 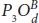 and
and  conformers take negative values for some ranges of angles, which lead to an auxetic behavior (see [79]). Moreover, in all the conformers, the Poisson ratio is lower than 0.5, which correspond to incompressible materials. Stress-strain responses, under the armchair and zigzag tensile strain, show that half oxidation leads to much higher critical points, compared to pure phosphorene. It is found that both the dangling and bridge structures resist to a large axial deformation up to 27%–39% along the AC-axis with respect to the ZZ one. More precisely, the maximum values coincide with the dangling structures P 4O U, P 2O D, and P 3O Uwhich can resist a tensile strain up to 30%, 33%, and 39%, respectively, showing a high flexibility in the armchair direction. This result is owing to the high buckled honeycomb structures that exhibit these configurations in this direction. The mechanical flexibility of half-oxidized phosphorene make these structures an ideal candidate for wearable optoelectronic devices.
conformers take negative values for some ranges of angles, which lead to an auxetic behavior (see [79]). Moreover, in all the conformers, the Poisson ratio is lower than 0.5, which correspond to incompressible materials. Stress-strain responses, under the armchair and zigzag tensile strain, show that half oxidation leads to much higher critical points, compared to pure phosphorene. It is found that both the dangling and bridge structures resist to a large axial deformation up to 27%–39% along the AC-axis with respect to the ZZ one. More precisely, the maximum values coincide with the dangling structures P 4O U, P 2O D, and P 3O Uwhich can resist a tensile strain up to 30%, 33%, and 39%, respectively, showing a high flexibility in the armchair direction. This result is owing to the high buckled honeycomb structures that exhibit these configurations in this direction. The mechanical flexibility of half-oxidized phosphorene make these structures an ideal candidate for wearable optoelectronic devices.
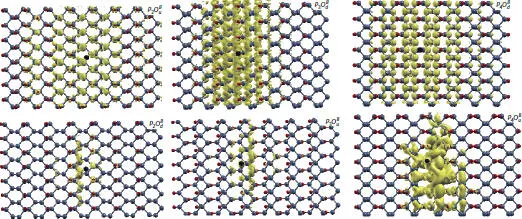
Figure 1.12 Excitons wave functions. Black balls represent the holes.
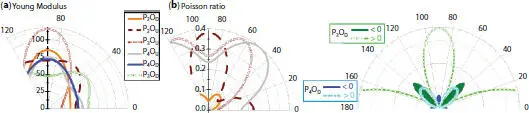
Figure 1.13 Part of polar plots of (a) Young modulus, (b) Poisson ratios.
Under half oxidation, the Debye temperature of phosphorene increases, with a maximum value reached in the ZZ-axis relative to AC. The high Debye temperature values indicate an important thermal conductivity in these new derivatives lattice lattice [79]. Furthermore, the curves describing the normal electrical polarization of the PO configurations in terms of applied strain are linear. With respect to pure phosphorene, the piezoelectric stress parameters increase under 50% oxidation while the piezoelectric strain coefficients d11 are three times lower than 2D BP [80].
When axial deformation is implemented, the electronic features of the POs become modulated. The band gap of dangling and bridge structures increases with low tensile strain, then it reduces to achieve a metallic state for large deformations. Besides, both groups of POs can maintain the semi-conductor behavior along the armchair direction for a strain ranging from 20% to 40% [81]. One can deduce that the adjustment of the phosphorene oxides features makes this class of materials potential candidates for advanced devices.
1.3.3 Surface Oxidation on Phosphorene
In contrast to the functionals H, F, and −OH which work like scissors by breaking down phosphorene into nanoribbons [82], a complete oxidation maintains the initial phosphorene configuration and shifts the lattice constants without breaking the Phosphorous bonds connecting the two P-half-layers, namely, the upper and lower ones [30].
1.3.3.1 Optoelectronic Features
At high concentration, oxidation leads to new derivatives of oxided phosphorene. As shown in Figure 1.14, up to fully oxidation, the interatomic P-P lengths increase to 2.32 and 2.37 Å while the direct gap located at point Γ reduces with respect to pure phosphorene. VBM is characterized by p y orbitals of P- and O-atoms, while the P-s and O-p zorbitals dominate the conduction band minimum (CBM) [30]. Both interstitial and dangling oxygen form no states in the middle of the gap, while the horizontal and diagonal oxygen introduce levels in the gap, which deals with a deep acceptor state at near the conduction band. Furthermore, the planar phosphorene oxides exhibit a monotonic increase to reach a maximum value with a deoxidation degree of 0.25, then start to decrease to attempt the value of 0.6 eV in a fully oxidized PO structure. For the tubular structure, the band gaps take the values from 0.4 (1.62) to 5.56 (7.78) eV at PBE (HSE level) [32]. Interestingly, the GW corrected band gap shows that the increasing oxygen coverage leads to an increase in the band energy from 4 eV to 10 eV, indicating that the VBM and CBM part become more localized [83].
Figure 1.14 (a) Top and (b) side views of phosphorene oxides PO.
The application of electric field reduces the gap energy of PO to a minimum of about 0.4 eV for a field E = 1.5 V/Å. The band gap fluctuates also from direct found for 100% to indirect for O-concentrations of 12.5%, 25%, and 50 %. Also, the work function in phosphorene increases linearly with the increased of the oxidation degree. The calculated values for PO 0.125, PO 0.25, and PO 0.5, are 4.9, 5.2, and 5.8, respectively, compared to PO that has 7.2 eV [30].
Under ambient conditions, phosphorene oxide is a stable material that did not exhibit any negative frequencies in its phonon dispersion curve [30]. Moreover, the simulation indicates that oxided structure is still robust and intact at low temperature, confirming its stability, while the material cut for large temperature values [84]. Unlike pure phosphorene, the phonon dispersion of PO exhibits three main regions as displayed in Figure 1.15b, namely, (i) the acoustic region, (ii) the middle region, and (iii) the high frequency range. Moreover, in contrast to the electron effective mass, the effective masses of holes are anisotrope [30].
Besides the band structure modification, the oxidation tunes also the optical features of phosphorene. In PO systems, the absorption spectrum reveals that the 1 st absorption peak is located at 2.7 eV, in P 4O 2, it is also found that both phosphorus and oxygen atoms contribute in the transition and extend the wavefunction along the AC-axis (see Figure 1.16a). Further, in the P 4O 10system, the absorption peak moves to high energy with a peak located at 7.0 eV. The wavefunction only localized on oxygen atoms adsorbed at the surface (see Figure 1.16b). This changes the binding energy E b from −1.4 eV to reach −3.0 eV for P 4O 2and P 4O 10. The electronic and optical band gap as well as the binding energy of P 4O 2and P 4O 10are very close to those of benzene [83].
Читать дальшеИнтервал:
Закладка:
Обсуждение, отзывы о книге «2D Monoelements» и просто собственные мнения читателей. Оставьте ваши комментарии, напишите, что Вы думаете о произведении, его смысле или главных героях. Укажите что конкретно понравилось, а что нет, и почему Вы так считаете.